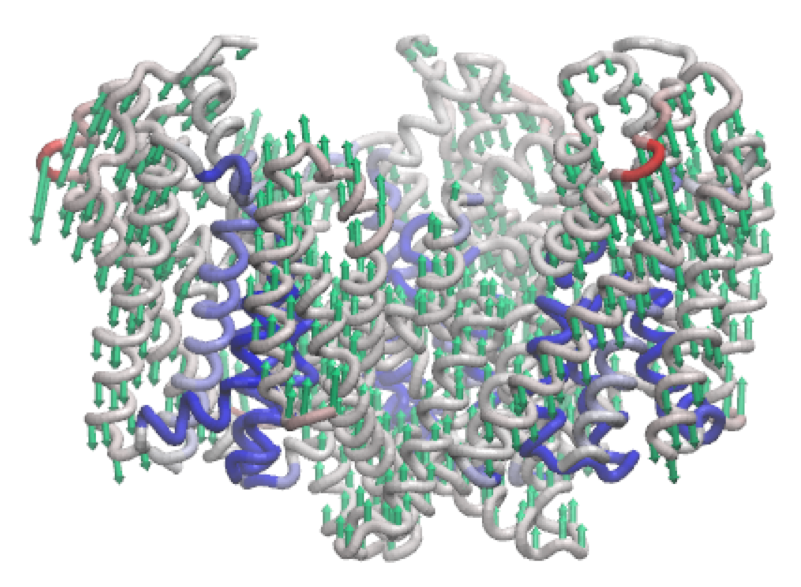
The third mode of the outward-facing structure moves all three transport domains simultaneously through the membrane in a 'lift-like' motion.
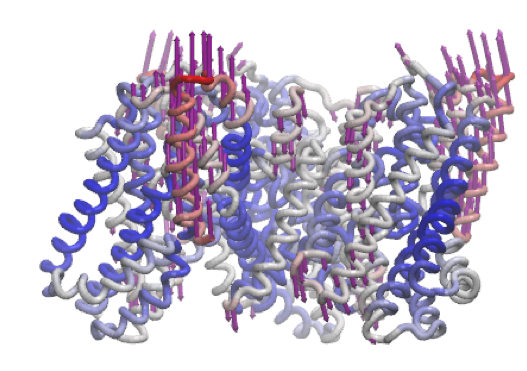 A similar motion is shown in mode 6 of the inward-facing structure.
A similar motion is shown in mode 6 of the inward-facing structure.
Here we will make use of ProDy’s membrane ANM capabilities to investigate the motions of a neurotransmitter transporter in the presence of the plasma membrane. The procedure is based on the methods described in:
Lezon TR, Bahar I. Constraints Imposed by the Membrane Selectively Guide the Alternating Access Dynamics of the Glutamate Transporter GltPh. Biophys J 2012 102 1331-1340.
You will need files containing the inward- and outward-facing structures of the glutamate transporter after insertion into the plasma membrane. It is obtained from the Orientations of Proteins in Membranes database.
Please ensure that you have downloaded and unzipped the tutorial materials from this link.
imANM relies on the Rotations and Translations of Blocks (RTB) method of reducing complexity within ENMs:
Tama F, Gadea FJ, Marques O, Sanejouand YH. Building-block approach for determining low-frequency normal modes of macromolecules. Proteins 2000 41 1-7.
We start by importing necessary modules:
from prody import *
from numpy import *
from matplotlib.pyplot import *
%matplotlib inline
confProDy(auto_show=False)
confProDy(auto_secondary=True)
@> ProDy is configured: auto_show=False @> ProDy is configured: auto_secondary=True
The imANM assumes that the membrane is normal to the z-axis, so it is important to use a structure that is properly aligned. The structure from the OPM database will work.
of_all = parsePDB('./membrane_anm_files/2NWL-opm.pdb') # Outward-facing structure
if_all = parsePDB('./membrane_anm_files/3KBC-opm.pdb') # Inward-facing structure
@> WARNING failed to parse beta-factor at line 8730 @> WARNING failed to parse beta-factor at line 8731 @> WARNING failed to parse beta-factor at line 8732 @> WARNING failed to parse beta-factor at line 8733 @> WARNING failed to parse beta-factor at line 8734 @> WARNING failed to parse beta-factor at line 8735 @> WARNING failed to parse beta-factor at line 8736 @> WARNING failed to parse beta-factor at line 8737 @> WARNING failed to parse beta-factor at line 8738 @> WARNING failed to parse beta-factor at line 8739 @> WARNING failed to parse beta-factor at line 8740 @> WARNING failed to parse beta-factor at line 8741 @> WARNING failed to parse beta-factor at line 8742 @> WARNING failed to parse beta-factor at line 8743 @> WARNING failed to parse beta-factor at line 8744 @> WARNING failed to parse beta-factor at line 8745 @> WARNING failed to parse beta-factor at line 8746 @> WARNING failed to parse beta-factor at line 8747 @> WARNING failed to parse beta-factor at line 8748 @> WARNING failed to parse beta-factor at line 8749 @> WARNING failed to parse beta-factor at line 8750 @> WARNING failed to parse beta-factor at line 8751 @> WARNING failed to parse beta-factor at line 8752 @> WARNING failed to parse beta-factor at line 8753 @> WARNING failed to parse beta-factor at line 8754 @> WARNING failed to parse beta-factor at line 8755 @> WARNING failed to parse beta-factor at line 8756 @> WARNING failed to parse beta-factor at line 8757 @> WARNING failed to parse beta-factor at line 8758 @> WARNING failed to parse beta-factor at line 8759 @> WARNING failed to parse beta-factor at line 8760 @> WARNING failed to parse beta-factor at line 8761 @> WARNING failed to parse beta-factor at line 8762 @> WARNING failed to parse beta-factor at line 8763 @> WARNING failed to parse beta-factor at line 8764 @> WARNING failed to parse beta-factor at line 8765 @> WARNING failed to parse beta-factor at line 8766 @> WARNING failed to parse beta-factor at line 8767 @> WARNING failed to parse beta-factor at line 8768 @> WARNING failed to parse beta-factor at line 8769 @> WARNING failed to parse beta-factor at line 8770 @> WARNING failed to parse beta-factor at line 8771 @> WARNING failed to parse beta-factor at line 8772 @> WARNING failed to parse beta-factor at line 8773 @> WARNING failed to parse beta-factor at line 8774 @> WARNING failed to parse beta-factor at line 8775 @> WARNING failed to parse beta-factor at line 8776 @> WARNING failed to parse beta-factor at line 8777 @> WARNING failed to parse beta-factor at line 8778 @> WARNING failed to parse beta-factor at line 8779 @> WARNING failed to parse beta-factor at line 8780 @> WARNING failed to parse beta-factor at line 8781 @> WARNING failed to parse beta-factor at line 8782 @> WARNING failed to parse beta-factor at line 8783 @> WARNING failed to parse beta-factor at line 8784 @> WARNING failed to parse beta-factor at line 8785 @> WARNING failed to parse beta-factor at line 8786 @> WARNING failed to parse beta-factor at line 8787 @> WARNING failed to parse beta-factor at line 8788 @> WARNING failed to parse beta-factor at line 8789 @> WARNING failed to parse beta-factor at line 8790 @> WARNING failed to parse beta-factor at line 8791 @> WARNING failed to parse beta-factor at line 8792 @> WARNING failed to parse beta-factor at line 8793 @> WARNING failed to parse beta-factor at line 8794 @> WARNING failed to parse beta-factor at line 8795 @> WARNING failed to parse beta-factor at line 8796 @> WARNING failed to parse beta-factor at line 8797 @> WARNING failed to parse beta-factor at line 8798 @> WARNING failed to parse beta-factor at line 8799 @> WARNING failed to parse beta-factor at line 8800 @> WARNING failed to parse beta-factor at line 8801 @> WARNING failed to parse beta-factor at line 8802 @> WARNING failed to parse beta-factor at line 8803 @> WARNING failed to parse beta-factor at line 8804 @> WARNING failed to parse beta-factor at line 8805 @> WARNING failed to parse beta-factor at line 8806 @> WARNING failed to parse beta-factor at line 8807 @> WARNING failed to parse beta-factor at line 8808 @> WARNING failed to parse beta-factor at line 8809 @> WARNING failed to parse beta-factor at line 8810 @> WARNING failed to parse beta-factor at line 8811 @> WARNING failed to parse beta-factor at line 8812 @> WARNING failed to parse beta-factor at line 8813 @> WARNING failed to parse beta-factor at line 8814 @> WARNING failed to parse beta-factor at line 8815 @> WARNING failed to parse beta-factor at line 8816 @> WARNING failed to parse beta-factor at line 8817 @> WARNING failed to parse beta-factor at line 8818 @> WARNING failed to parse beta-factor at line 8819 @> WARNING failed to parse beta-factor at line 8820 @> WARNING failed to parse beta-factor at line 8821 @> WARNING failed to parse beta-factor at line 8822 @> WARNING failed to parse beta-factor at line 8823 @> WARNING failed to parse beta-factor at line 8824 @> WARNING failed to parse beta-factor at line 8825 @> WARNING failed to parse beta-factor at line 8826 @> WARNING failed to parse beta-factor at line 8827 @> WARNING failed to parse beta-factor at line 8828 @> WARNING failed to parse beta-factor at line 8829 @> WARNING failed to parse beta-factor at line 8830 @> WARNING failed to parse beta-factor at line 8831 @> WARNING failed to parse beta-factor at line 8832 @> WARNING failed to parse beta-factor at line 8833 @> WARNING failed to parse beta-factor at line 8834 @> WARNING failed to parse beta-factor at line 8835 @> WARNING failed to parse beta-factor at line 8836 @> WARNING failed to parse beta-factor at line 8837 @> WARNING failed to parse beta-factor at line 8838 @> WARNING failed to parse beta-factor at line 8839 @> WARNING failed to parse beta-factor at line 8840 @> WARNING failed to parse beta-factor at line 8841 @> WARNING failed to parse beta-factor at line 8842 @> WARNING failed to parse beta-factor at line 8843 @> WARNING failed to parse beta-factor at line 8844 @> WARNING failed to parse beta-factor at line 8845 @> WARNING failed to parse beta-factor at line 8846 @> WARNING failed to parse beta-factor at line 8847 @> WARNING failed to parse beta-factor at line 8848 @> WARNING failed to parse beta-factor at line 8849 @> WARNING failed to parse beta-factor at line 8850 @> WARNING failed to parse beta-factor at line 8851 @> WARNING failed to parse beta-factor at line 8852 @> WARNING failed to parse beta-factor at line 8853 @> WARNING failed to parse beta-factor at line 8854 @> WARNING failed to parse beta-factor at line 8855 @> WARNING failed to parse beta-factor at line 8856 @> WARNING failed to parse beta-factor at line 8857 @> WARNING failed to parse beta-factor at line 8858 @> WARNING failed to parse beta-factor at line 8859 @> WARNING failed to parse beta-factor at line 8860 @> WARNING failed to parse beta-factor at line 8861 @> WARNING failed to parse beta-factor at line 8862 @> WARNING failed to parse beta-factor at line 8863 @> WARNING failed to parse beta-factor at line 8864 @> WARNING failed to parse beta-factor at line 8865 @> WARNING failed to parse beta-factor at line 8866 @> WARNING failed to parse beta-factor at line 8867 @> WARNING failed to parse beta-factor at line 8868 @> WARNING failed to parse beta-factor at line 8869 @> WARNING failed to parse beta-factor at line 8870 @> WARNING failed to parse beta-factor at line 8871 @> WARNING failed to parse beta-factor at line 8872 @> WARNING failed to parse beta-factor at line 8873 @> WARNING failed to parse beta-factor at line 8874 @> WARNING failed to parse beta-factor at line 8875 @> WARNING failed to parse beta-factor at line 8876 @> WARNING failed to parse beta-factor at line 8877 @> WARNING failed to parse beta-factor at line 8878 @> WARNING failed to parse beta-factor at line 8879 @> WARNING failed to parse beta-factor at line 8880 @> WARNING failed to parse beta-factor at line 8881 @> WARNING failed to parse beta-factor at line 8882 @> WARNING failed to parse beta-factor at line 8883 @> WARNING failed to parse beta-factor at line 8884 @> WARNING failed to parse beta-factor at line 8885 @> WARNING failed to parse beta-factor at line 8886 @> WARNING failed to parse beta-factor at line 8887 @> WARNING failed to parse beta-factor at line 8888 @> WARNING failed to parse beta-factor at line 8889 @> WARNING failed to parse beta-factor at line 8890 @> WARNING failed to parse beta-factor at line 8891 @> WARNING failed to parse beta-factor at line 8892 @> WARNING failed to parse beta-factor at line 8893 @> WARNING failed to parse beta-factor at line 8894 @> WARNING failed to parse beta-factor at line 8895 @> WARNING failed to parse beta-factor at line 8896 @> WARNING failed to parse beta-factor at line 8897 @> WARNING failed to parse beta-factor at line 8898 @> WARNING failed to parse beta-factor at line 8899 @> WARNING failed to parse beta-factor at line 8900 @> WARNING failed to parse beta-factor at line 8901 @> WARNING failed to parse beta-factor at line 8902 @> WARNING failed to parse beta-factor at line 8903 @> WARNING failed to parse beta-factor at line 8904 @> WARNING failed to parse beta-factor at line 8905 @> WARNING failed to parse beta-factor at line 8906 @> WARNING failed to parse beta-factor at line 8907 @> WARNING failed to parse beta-factor at line 8908 @> WARNING failed to parse beta-factor at line 8909 @> WARNING failed to parse beta-factor at line 8910 @> WARNING failed to parse beta-factor at line 8911 @> WARNING failed to parse beta-factor at line 8912 @> WARNING failed to parse beta-factor at line 8913 @> WARNING failed to parse beta-factor at line 8914 @> WARNING failed to parse beta-factor at line 8915 @> WARNING failed to parse beta-factor at line 8916 @> WARNING failed to parse beta-factor at line 8917 @> WARNING failed to parse beta-factor at line 8918 @> WARNING failed to parse beta-factor at line 8919 @> WARNING failed to parse beta-factor at line 8920 @> WARNING failed to parse beta-factor at line 8921 @> WARNING failed to parse beta-factor at line 8922 @> WARNING failed to parse beta-factor at line 8923 @> WARNING failed to parse beta-factor at line 8924 @> WARNING failed to parse beta-factor at line 8925 @> WARNING failed to parse beta-factor at line 8926 @> WARNING failed to parse beta-factor at line 8927 @> WARNING failed to parse beta-factor at line 8928 @> WARNING failed to parse beta-factor at line 8929 @> WARNING failed to parse beta-factor at line 8930 @> WARNING failed to parse beta-factor at line 8931 @> WARNING failed to parse beta-factor at line 8932 @> WARNING failed to parse beta-factor at line 8933 @> WARNING failed to parse beta-factor at line 8934 @> WARNING failed to parse beta-factor at line 8935 @> WARNING failed to parse beta-factor at line 8936 @> WARNING failed to parse beta-factor at line 8937 @> WARNING failed to parse beta-factor at line 8938 @> WARNING failed to parse beta-factor at line 8939 @> WARNING failed to parse beta-factor at line 8940 @> WARNING failed to parse beta-factor at line 8941 @> WARNING failed to parse beta-factor at line 8942 @> WARNING failed to parse beta-factor at line 8943 @> WARNING failed to parse beta-factor at line 8944 @> WARNING failed to parse beta-factor at line 8945 @> WARNING failed to parse beta-factor at line 8946 @> WARNING failed to parse beta-factor at line 8947 @> WARNING failed to parse beta-factor at line 8948 @> WARNING failed to parse beta-factor at line 8949 @> WARNING failed to parse beta-factor at line 8950 @> WARNING failed to parse beta-factor at line 8951 @> WARNING failed to parse beta-factor at line 8952 @> WARNING failed to parse beta-factor at line 8953 @> WARNING failed to parse beta-factor at line 8954 @> WARNING failed to parse beta-factor at line 8955 @> WARNING failed to parse beta-factor at line 8956 @> WARNING failed to parse beta-factor at line 8957 @> WARNING failed to parse beta-factor at line 8958 @> WARNING failed to parse beta-factor at line 8959 @> WARNING failed to parse beta-factor at line 8960 @> WARNING failed to parse beta-factor at line 8961 @> WARNING failed to parse beta-factor at line 8962 @> WARNING failed to parse beta-factor at line 8963 @> WARNING failed to parse beta-factor at line 8964 @> WARNING failed to parse beta-factor at line 8965 @> WARNING failed to parse beta-factor at line 8966 @> WARNING failed to parse beta-factor at line 8967 @> WARNING failed to parse beta-factor at line 8968 @> WARNING failed to parse beta-factor at line 8969 @> WARNING failed to parse beta-factor at line 8970 @> WARNING failed to parse beta-factor at line 8971 @> WARNING failed to parse beta-factor at line 8972 @> WARNING failed to parse beta-factor at line 8973 @> WARNING failed to parse beta-factor at line 8974 @> WARNING failed to parse beta-factor at line 8975 @> WARNING failed to parse beta-factor at line 8976 @> WARNING failed to parse beta-factor at line 8977 @> WARNING failed to parse beta-factor at line 8978 @> WARNING failed to parse beta-factor at line 8979 @> WARNING failed to parse beta-factor at line 8980 @> WARNING failed to parse beta-factor at line 8981 @> WARNING failed to parse beta-factor at line 8982 @> WARNING failed to parse beta-factor at line 8983 @> WARNING failed to parse beta-factor at line 8984 @> WARNING failed to parse beta-factor at line 8985 @> WARNING failed to parse beta-factor at line 8986 @> WARNING failed to parse beta-factor at line 8987 @> WARNING failed to parse beta-factor at line 8988 @> WARNING failed to parse beta-factor at line 8989 @> WARNING failed to parse beta-factor at line 8990 @> WARNING failed to parse beta-factor at line 8991 @> WARNING failed to parse beta-factor at line 8992 @> WARNING failed to parse beta-factor at line 8993 @> WARNING failed to parse beta-factor at line 8994 @> WARNING failed to parse beta-factor at line 8995 @> WARNING failed to parse beta-factor at line 8996 @> WARNING failed to parse beta-factor at line 8997 @> WARNING failed to parse beta-factor at line 8998 @> WARNING failed to parse beta-factor at line 8999 @> WARNING failed to parse beta-factor at line 9000 @> WARNING failed to parse beta-factor at line 9001 @> WARNING failed to parse beta-factor at line 9002 @> WARNING failed to parse beta-factor at line 9003 @> WARNING failed to parse beta-factor at line 9004 @> WARNING failed to parse beta-factor at line 9005 @> WARNING failed to parse beta-factor at line 9006 @> WARNING failed to parse beta-factor at line 9007 @> WARNING failed to parse beta-factor at line 9008 @> WARNING failed to parse beta-factor at line 9009 @> WARNING failed to parse beta-factor at line 9010 @> WARNING failed to parse beta-factor at line 9011 @> WARNING failed to parse beta-factor at line 9012 @> WARNING failed to parse beta-factor at line 9013 @> WARNING failed to parse beta-factor at line 9014 @> WARNING failed to parse beta-factor at line 9015 @> WARNING failed to parse beta-factor at line 9016 @> WARNING failed to parse beta-factor at line 9017 @> WARNING failed to parse beta-factor at line 9018 @> WARNING failed to parse beta-factor at line 9019 @> WARNING failed to parse beta-factor at line 9020 @> WARNING failed to parse beta-factor at line 9021 @> WARNING failed to parse beta-factor at line 9022 @> WARNING failed to parse beta-factor at line 9023 @> WARNING failed to parse beta-factor at line 9024 @> WARNING failed to parse beta-factor at line 9025 @> WARNING failed to parse beta-factor at line 9026 @> WARNING failed to parse beta-factor at line 9027 @> WARNING failed to parse beta-factor at line 9028 @> WARNING failed to parse beta-factor at line 9029 @> WARNING failed to parse beta-factor at line 9030 @> WARNING failed to parse beta-factor at line 9031 @> WARNING failed to parse beta-factor at line 9032 @> WARNING failed to parse beta-factor at line 9033 @> WARNING failed to parse beta-factor at line 9034 @> WARNING failed to parse beta-factor at line 9035 @> WARNING failed to parse beta-factor at line 9036 @> WARNING failed to parse beta-factor at line 9037 @> WARNING failed to parse beta-factor at line 9038 @> WARNING failed to parse beta-factor at line 9039 @> WARNING failed to parse beta-factor at line 9040 @> WARNING failed to parse beta-factor at line 9041 @> WARNING failed to parse beta-factor at line 9042 @> WARNING failed to parse beta-factor at line 9043 @> WARNING failed to parse beta-factor at line 9044 @> WARNING failed to parse beta-factor at line 9045 @> WARNING failed to parse beta-factor at line 9046 @> WARNING failed to parse beta-factor at line 9047 @> WARNING failed to parse beta-factor at line 9048 @> WARNING failed to parse beta-factor at line 9049 @> WARNING failed to parse beta-factor at line 9050 @> WARNING failed to parse beta-factor at line 9051 @> WARNING failed to parse beta-factor at line 9052 @> WARNING failed to parse beta-factor at line 9053 @> WARNING failed to parse beta-factor at line 9054 @> WARNING failed to parse beta-factor at line 9055 @> WARNING failed to parse beta-factor at line 9056 @> WARNING failed to parse beta-factor at line 9057 @> WARNING failed to parse beta-factor at line 9058 @> WARNING failed to parse beta-factor at line 9059 @> WARNING failed to parse beta-factor at line 9060 @> WARNING failed to parse beta-factor at line 9061 @> WARNING failed to parse beta-factor at line 9062 @> WARNING failed to parse beta-factor at line 9063 @> WARNING failed to parse beta-factor at line 9064 @> WARNING failed to parse beta-factor at line 9065 @> WARNING failed to parse beta-factor at line 9066 @> WARNING failed to parse beta-factor at line 9067 @> WARNING failed to parse beta-factor at line 9068 @> WARNING failed to parse beta-factor at line 9069 @> WARNING failed to parse beta-factor at line 9070 @> WARNING failed to parse beta-factor at line 9071 @> WARNING failed to parse beta-factor at line 9072 @> WARNING failed to parse beta-factor at line 9073 @> WARNING failed to parse beta-factor at line 9074 @> WARNING failed to parse beta-factor at line 9075 @> WARNING failed to parse beta-factor at line 9076 @> WARNING failed to parse beta-factor at line 9077 @> WARNING failed to parse beta-factor at line 9078 @> WARNING failed to parse beta-factor at line 9079 @> WARNING failed to parse beta-factor at line 9080 @> WARNING failed to parse beta-factor at line 9081 @> WARNING failed to parse beta-factor at line 9082 @> WARNING failed to parse beta-factor at line 9083 @> WARNING failed to parse beta-factor at line 9084 @> WARNING failed to parse beta-factor at line 9085 @> WARNING failed to parse beta-factor at line 9086 @> WARNING failed to parse beta-factor at line 9087 @> WARNING failed to parse beta-factor at line 9088 @> WARNING failed to parse beta-factor at line 9089 @> WARNING failed to parse beta-factor at line 9090 @> WARNING failed to parse beta-factor at line 9091 @> WARNING failed to parse beta-factor at line 9092 @> WARNING failed to parse beta-factor at line 9093 @> WARNING failed to parse beta-factor at line 9094 @> WARNING failed to parse beta-factor at line 9095 @> WARNING failed to parse beta-factor at line 9096 @> WARNING failed to parse beta-factor at line 9097 @> WARNING failed to parse beta-factor at line 9098 @> WARNING failed to parse beta-factor at line 9099 @> WARNING failed to parse beta-factor at line 9100 @> WARNING failed to parse beta-factor at line 9101 @> WARNING failed to parse beta-factor at line 9102 @> WARNING failed to parse beta-factor at line 9103 @> WARNING failed to parse beta-factor at line 9104 @> WARNING failed to parse beta-factor at line 9105 @> WARNING failed to parse beta-factor at line 9106 @> WARNING failed to parse beta-factor at line 9107 @> WARNING failed to parse beta-factor at line 9108 @> WARNING failed to parse beta-factor at line 9109 @> WARNING failed to parse beta-factor at line 9110 @> WARNING failed to parse beta-factor at line 9111 @> WARNING failed to parse beta-factor at line 9112 @> WARNING failed to parse beta-factor at line 9113 @> WARNING failed to parse beta-factor at line 9114 @> WARNING failed to parse beta-factor at line 9115 @> WARNING failed to parse beta-factor at line 9116 @> WARNING failed to parse beta-factor at line 9117 @> WARNING failed to parse beta-factor at line 9118 @> WARNING failed to parse beta-factor at line 9119 @> WARNING failed to parse beta-factor at line 9120 @> WARNING failed to parse beta-factor at line 9121 @> WARNING failed to parse beta-factor at line 9122 @> WARNING failed to parse beta-factor at line 9123 @> WARNING failed to parse beta-factor at line 9124 @> WARNING failed to parse beta-factor at line 9125 @> WARNING failed to parse beta-factor at line 9126 @> WARNING failed to parse beta-factor at line 9127 @> WARNING failed to parse beta-factor at line 9128 @> WARNING failed to parse beta-factor at line 9129 @> WARNING failed to parse beta-factor at line 9130 @> WARNING failed to parse beta-factor at line 9131 @> WARNING failed to parse beta-factor at line 9132 @> WARNING failed to parse beta-factor at line 9133 @> WARNING failed to parse beta-factor at line 9134 @> WARNING failed to parse beta-factor at line 9135 @> WARNING failed to parse beta-factor at line 9136 @> WARNING failed to parse beta-factor at line 9137 @> WARNING failed to parse beta-factor at line 9138 @> WARNING failed to parse beta-factor at line 9139 @> WARNING failed to parse beta-factor at line 9140 @> WARNING failed to parse beta-factor at line 9141 @> WARNING failed to parse beta-factor at line 9142 @> WARNING failed to parse beta-factor at line 9143 @> WARNING failed to parse beta-factor at line 9144 @> WARNING failed to parse beta-factor at line 9145 @> WARNING failed to parse beta-factor at line 9146 @> WARNING failed to parse beta-factor at line 9147 @> WARNING failed to parse beta-factor at line 9148 @> WARNING failed to parse beta-factor at line 9149 @> WARNING failed to parse beta-factor at line 9150 @> WARNING failed to parse beta-factor at line 9151 @> WARNING failed to parse beta-factor at line 9152 @> WARNING failed to parse beta-factor at line 9153 @> WARNING failed to parse beta-factor at line 9154 @> WARNING failed to parse beta-factor at line 9155 @> WARNING failed to parse beta-factor at line 9156 @> WARNING failed to parse beta-factor at line 9157 @> WARNING failed to parse beta-factor at line 9158 @> WARNING failed to parse beta-factor at line 9159 @> WARNING failed to parse beta-factor at line 9160 @> WARNING failed to parse beta-factor at line 9161 @> WARNING failed to parse beta-factor at line 9162 @> WARNING failed to parse beta-factor at line 9163 @> WARNING failed to parse beta-factor at line 9164 @> WARNING failed to parse beta-factor at line 9165 @> WARNING failed to parse beta-factor at line 9166 @> WARNING failed to parse beta-factor at line 9167 @> WARNING failed to parse beta-factor at line 9168 @> WARNING failed to parse beta-factor at line 9169 @> WARNING failed to parse beta-factor at line 9170 @> WARNING failed to parse beta-factor at line 9171 @> WARNING failed to parse beta-factor at line 9172 @> WARNING failed to parse beta-factor at line 9173 @> WARNING failed to parse beta-factor at line 9174 @> WARNING failed to parse beta-factor at line 9175 @> WARNING failed to parse beta-factor at line 9176 @> WARNING failed to parse beta-factor at line 9177 @> WARNING failed to parse beta-factor at line 9178 @> WARNING failed to parse beta-factor at line 9179 @> WARNING failed to parse beta-factor at line 9180 @> WARNING failed to parse beta-factor at line 9181 @> WARNING failed to parse beta-factor at line 9182 @> WARNING failed to parse beta-factor at line 9183 @> WARNING failed to parse beta-factor at line 9184 @> WARNING failed to parse beta-factor at line 9185 @> WARNING failed to parse beta-factor at line 9186 @> WARNING failed to parse beta-factor at line 9187 @> WARNING failed to parse beta-factor at line 9188 @> WARNING failed to parse beta-factor at line 9189 @> WARNING failed to parse beta-factor at line 9190 @> WARNING failed to parse beta-factor at line 9191 @> WARNING failed to parse beta-factor at line 9192 @> WARNING failed to parse beta-factor at line 9193 @> WARNING failed to parse beta-factor at line 9194 @> WARNING failed to parse beta-factor at line 9195 @> WARNING failed to parse beta-factor at line 9196 @> WARNING failed to parse beta-factor at line 9197 @> WARNING failed to parse beta-factor at line 9198 @> WARNING failed to parse beta-factor at line 9199 @> WARNING failed to parse beta-factor at line 9200 @> WARNING failed to parse beta-factor at line 9201 @> WARNING failed to parse beta-factor at line 9202 @> WARNING failed to parse beta-factor at line 9203 @> WARNING failed to parse beta-factor at line 9204 @> WARNING failed to parse beta-factor at line 9205 @> WARNING failed to parse beta-factor at line 9206 @> WARNING failed to parse beta-factor at line 9207 @> WARNING failed to parse beta-factor at line 9208 @> WARNING failed to parse beta-factor at line 9209 @> WARNING failed to parse beta-factor at line 9210 @> WARNING failed to parse beta-factor at line 9211 @> WARNING failed to parse beta-factor at line 9212 @> WARNING failed to parse beta-factor at line 9213 @> WARNING failed to parse beta-factor at line 9214 @> WARNING failed to parse beta-factor at line 9215 @> WARNING failed to parse beta-factor at line 9216 @> WARNING failed to parse beta-factor at line 9217 @> WARNING failed to parse beta-factor at line 9218 @> WARNING failed to parse beta-factor at line 9219 @> WARNING failed to parse beta-factor at line 9220 @> WARNING failed to parse beta-factor at line 9221 @> WARNING failed to parse beta-factor at line 9222 @> WARNING failed to parse beta-factor at line 9223 @> WARNING failed to parse beta-factor at line 9224 @> WARNING failed to parse beta-factor at line 9225 @> WARNING failed to parse beta-factor at line 9226 @> WARNING failed to parse beta-factor at line 9227 @> WARNING failed to parse beta-factor at line 9228 @> WARNING failed to parse beta-factor at line 9229 @> WARNING failed to parse beta-factor at line 9230 @> WARNING failed to parse beta-factor at line 9231 @> WARNING failed to parse beta-factor at line 9232 @> WARNING failed to parse beta-factor at line 9233 @> WARNING failed to parse beta-factor at line 9234 @> WARNING failed to parse beta-factor at line 9235 @> WARNING failed to parse beta-factor at line 9236 @> WARNING failed to parse beta-factor at line 9237 @> WARNING failed to parse beta-factor at line 9238 @> WARNING failed to parse beta-factor at line 9239 @> WARNING failed to parse beta-factor at line 9240 @> WARNING failed to parse beta-factor at line 9241 @> WARNING failed to parse beta-factor at line 9242 @> WARNING failed to parse beta-factor at line 9243 @> WARNING failed to parse beta-factor at line 9244 @> WARNING failed to parse beta-factor at line 9245 @> WARNING failed to parse beta-factor at line 9246 @> WARNING failed to parse beta-factor at line 9247 @> WARNING failed to parse beta-factor at line 9248 @> WARNING failed to parse beta-factor at line 9249 @> WARNING failed to parse beta-factor at line 9250 @> WARNING failed to parse beta-factor at line 9251 @> WARNING failed to parse beta-factor at line 9252 @> WARNING failed to parse beta-factor at line 9253 @> WARNING failed to parse beta-factor at line 9254 @> WARNING failed to parse beta-factor at line 9255 @> WARNING failed to parse beta-factor at line 9256 @> WARNING failed to parse beta-factor at line 9257 @> WARNING failed to parse beta-factor at line 9258 @> WARNING failed to parse beta-factor at line 9259 @> WARNING failed to parse beta-factor at line 9260 @> WARNING failed to parse beta-factor at line 9261 @> WARNING failed to parse beta-factor at line 9262 @> WARNING failed to parse beta-factor at line 9263 @> WARNING failed to parse beta-factor at line 9264 @> WARNING failed to parse beta-factor at line 9265 @> WARNING failed to parse beta-factor at line 9266 @> WARNING failed to parse beta-factor at line 9267 @> WARNING failed to parse beta-factor at line 9268 @> WARNING failed to parse beta-factor at line 9269 @> WARNING failed to parse beta-factor at line 9270 @> WARNING failed to parse beta-factor at line 9271 @> WARNING failed to parse beta-factor at line 9272 @> WARNING failed to parse beta-factor at line 9273 @> WARNING failed to parse beta-factor at line 9274 @> WARNING failed to parse beta-factor at line 9275 @> WARNING failed to parse beta-factor at line 9276 @> WARNING failed to parse beta-factor at line 9277 @> WARNING failed to parse beta-factor at line 9278 @> WARNING failed to parse beta-factor at line 9279 @> WARNING failed to parse beta-factor at line 9280 @> WARNING failed to parse beta-factor at line 9281 @> WARNING failed to parse beta-factor at line 9282 @> WARNING failed to parse beta-factor at line 9283 @> WARNING failed to parse beta-factor at line 9284 @> WARNING failed to parse beta-factor at line 9285 @> WARNING failed to parse beta-factor at line 9286 @> WARNING failed to parse beta-factor at line 9287 @> WARNING failed to parse beta-factor at line 9288 @> WARNING failed to parse beta-factor at line 9289 @> WARNING failed to parse beta-factor at line 9290 @> WARNING failed to parse beta-factor at line 9291 @> WARNING failed to parse beta-factor at line 9292 @> WARNING failed to parse beta-factor at line 9293 @> WARNING failed to parse beta-factor at line 9294 @> WARNING failed to parse beta-factor at line 9295 @> WARNING failed to parse beta-factor at line 9296 @> WARNING failed to parse beta-factor at line 9297 @> WARNING failed to parse beta-factor at line 9298 @> WARNING failed to parse beta-factor at line 9299 @> WARNING failed to parse beta-factor at line 9300 @> WARNING failed to parse beta-factor at line 9301 @> WARNING failed to parse beta-factor at line 9302 @> WARNING failed to parse beta-factor at line 9303 @> WARNING failed to parse beta-factor at line 9304 @> WARNING failed to parse beta-factor at line 9305 @> WARNING failed to parse beta-factor at line 9306 @> WARNING failed to parse beta-factor at line 9307 @> WARNING failed to parse beta-factor at line 9308 @> WARNING failed to parse beta-factor at line 9309 @> WARNING failed to parse beta-factor at line 9310 @> WARNING failed to parse beta-factor at line 9311 @> WARNING failed to parse beta-factor at line 9312 @> WARNING failed to parse beta-factor at line 9313 @> WARNING failed to parse beta-factor at line 9314 @> WARNING failed to parse beta-factor at line 9315 @> WARNING failed to parse beta-factor at line 9316 @> WARNING failed to parse beta-factor at line 9317 @> WARNING failed to parse beta-factor at line 9318 @> WARNING failed to parse beta-factor at line 9319 @> WARNING failed to parse beta-factor at line 9320 @> WARNING failed to parse beta-factor at line 9321 @> WARNING failed to parse beta-factor at line 9322 @> WARNING failed to parse beta-factor at line 9323 @> WARNING failed to parse beta-factor at line 9324 @> WARNING failed to parse beta-factor at line 9325 @> WARNING failed to parse beta-factor at line 9326 @> WARNING failed to parse beta-factor at line 9327 @> WARNING failed to parse beta-factor at line 9328 @> WARNING failed to parse beta-factor at line 9329 @> WARNING failed to parse beta-factor at line 9330 @> WARNING failed to parse beta-factor at line 9331 @> WARNING failed to parse beta-factor at line 9332 @> WARNING failed to parse beta-factor at line 9333 @> WARNING failed to parse beta-factor at line 9334 @> WARNING failed to parse beta-factor at line 9335 @> WARNING failed to parse beta-factor at line 9336 @> WARNING failed to parse beta-factor at line 9337 @> WARNING failed to parse beta-factor at line 9338 @> WARNING failed to parse beta-factor at line 9339 @> WARNING failed to parse beta-factor at line 9340 @> WARNING failed to parse beta-factor at line 9341 @> WARNING failed to parse beta-factor at line 9342 @> WARNING failed to parse beta-factor at line 9343 @> WARNING failed to parse beta-factor at line 9344 @> WARNING failed to parse beta-factor at line 9345 @> WARNING failed to parse beta-factor at line 9346 @> WARNING failed to parse beta-factor at line 9347 @> WARNING failed to parse beta-factor at line 9348 @> WARNING failed to parse beta-factor at line 9349 @> WARNING failed to parse beta-factor at line 9350 @> WARNING failed to parse beta-factor at line 9351 @> WARNING failed to parse beta-factor at line 9352 @> WARNING failed to parse beta-factor at line 9353 @> WARNING failed to parse beta-factor at line 9354 @> WARNING failed to parse beta-factor at line 9355 @> WARNING failed to parse beta-factor at line 9356 @> WARNING failed to parse beta-factor at line 9357 @> WARNING failed to parse beta-factor at line 9358 @> WARNING failed to parse beta-factor at line 9359 @> WARNING failed to parse beta-factor at line 9360 @> WARNING failed to parse beta-factor at line 9361 @> WARNING failed to parse beta-factor at line 9362 @> WARNING failed to parse beta-factor at line 9363 @> WARNING failed to parse beta-factor at line 9364 @> WARNING failed to parse beta-factor at line 9365 @> WARNING failed to parse beta-factor at line 9366 @> WARNING failed to parse beta-factor at line 9367 @> WARNING failed to parse beta-factor at line 9368 @> WARNING failed to parse beta-factor at line 9369 @> WARNING failed to parse beta-factor at line 9370 @> WARNING failed to parse beta-factor at line 9371 @> WARNING failed to parse beta-factor at line 9372 @> WARNING failed to parse beta-factor at line 9373 @> WARNING failed to parse beta-factor at line 9374 @> WARNING failed to parse beta-factor at line 9375 @> WARNING failed to parse beta-factor at line 9376 @> WARNING failed to parse beta-factor at line 9377 @> WARNING failed to parse beta-factor at line 9378 @> WARNING failed to parse beta-factor at line 9379 @> WARNING failed to parse beta-factor at line 9380 @> WARNING failed to parse beta-factor at line 9381 @> WARNING failed to parse beta-factor at line 9382 @> WARNING failed to parse beta-factor at line 9383 @> WARNING failed to parse beta-factor at line 9384 @> WARNING failed to parse beta-factor at line 9385 @> WARNING failed to parse beta-factor at line 9386 @> WARNING failed to parse beta-factor at line 9387 @> WARNING failed to parse beta-factor at line 9388 @> WARNING failed to parse beta-factor at line 9389 @> WARNING failed to parse beta-factor at line 9390 @> WARNING failed to parse beta-factor at line 9391 @> WARNING failed to parse beta-factor at line 9392 @> WARNING failed to parse beta-factor at line 9393 @> WARNING failed to parse beta-factor at line 9394 @> WARNING failed to parse beta-factor at line 9395 @> WARNING failed to parse beta-factor at line 9396 @> WARNING failed to parse beta-factor at line 9397 @> WARNING failed to parse beta-factor at line 9398 @> WARNING failed to parse beta-factor at line 9399 @> WARNING failed to parse beta-factor at line 9400 @> WARNING failed to parse beta-factor at line 9401 @> WARNING failed to parse beta-factor at line 9402 @> WARNING failed to parse beta-factor at line 9403 @> WARNING failed to parse beta-factor at line 9404 @> WARNING failed to parse beta-factor at line 9405 @> WARNING failed to parse beta-factor at line 9406 @> WARNING failed to parse beta-factor at line 9407 @> WARNING failed to parse beta-factor at line 9408 @> WARNING failed to parse beta-factor at line 9409 @> WARNING failed to parse beta-factor at line 9410 @> WARNING failed to parse beta-factor at line 9411 @> WARNING failed to parse beta-factor at line 9412 @> WARNING failed to parse beta-factor at line 9413 @> WARNING failed to parse beta-factor at line 9414 @> WARNING failed to parse beta-factor at line 9415 @> WARNING failed to parse beta-factor at line 9416 @> WARNING failed to parse beta-factor at line 9417 @> WARNING failed to parse beta-factor at line 9418 @> WARNING failed to parse beta-factor at line 9419 @> WARNING failed to parse beta-factor at line 9420 @> WARNING failed to parse beta-factor at line 9421 @> WARNING failed to parse beta-factor at line 9422 @> WARNING failed to parse beta-factor at line 9423 @> WARNING failed to parse beta-factor at line 9424 @> WARNING failed to parse beta-factor at line 9425 @> WARNING failed to parse beta-factor at line 9426 @> WARNING failed to parse beta-factor at line 9427 @> WARNING failed to parse beta-factor at line 9428 @> WARNING failed to parse beta-factor at line 9429 @> WARNING failed to parse beta-factor at line 9430 @> WARNING failed to parse beta-factor at line 9431 @> WARNING failed to parse beta-factor at line 9432 @> WARNING failed to parse beta-factor at line 9433 @> WARNING failed to parse beta-factor at line 9434 @> WARNING failed to parse beta-factor at line 9435 @> WARNING failed to parse beta-factor at line 9436 @> WARNING failed to parse beta-factor at line 9437 @> WARNING failed to parse beta-factor at line 9438 @> WARNING failed to parse beta-factor at line 9439 @> WARNING failed to parse beta-factor at line 9440 @> WARNING failed to parse beta-factor at line 9441 @> WARNING failed to parse beta-factor at line 9442 @> WARNING failed to parse beta-factor at line 9443 @> WARNING failed to parse beta-factor at line 9444 @> WARNING failed to parse beta-factor at line 9445 @> WARNING failed to parse beta-factor at line 9446 @> WARNING failed to parse beta-factor at line 9447 @> WARNING failed to parse beta-factor at line 9448 @> WARNING failed to parse beta-factor at line 9449 @> WARNING failed to parse beta-factor at line 9450 @> WARNING failed to parse beta-factor at line 9451 @> WARNING failed to parse beta-factor at line 9452 @> WARNING failed to parse beta-factor at line 9453 @> WARNING failed to parse beta-factor at line 9454 @> WARNING failed to parse beta-factor at line 9455 @> WARNING failed to parse beta-factor at line 9456 @> WARNING failed to parse beta-factor at line 9457 @> WARNING failed to parse beta-factor at line 9458 @> WARNING failed to parse beta-factor at line 9459 @> WARNING failed to parse beta-factor at line 9460 @> WARNING failed to parse beta-factor at line 9461 @> WARNING failed to parse beta-factor at line 9462 @> WARNING failed to parse beta-factor at line 9463 @> WARNING failed to parse beta-factor at line 9464 @> WARNING failed to parse beta-factor at line 9465 @> WARNING failed to parse beta-factor at line 9466 @> WARNING failed to parse beta-factor at line 9467 @> WARNING failed to parse beta-factor at line 9468 @> WARNING failed to parse beta-factor at line 9469 @> WARNING failed to parse beta-factor at line 9470 @> WARNING failed to parse beta-factor at line 9471 @> WARNING failed to parse beta-factor at line 9472 @> WARNING failed to parse beta-factor at line 9473 @> WARNING failed to parse beta-factor at line 9474 @> WARNING failed to parse beta-factor at line 9475 @> WARNING failed to parse beta-factor at line 9476 @> WARNING failed to parse beta-factor at line 9477 @> WARNING failed to parse beta-factor at line 9478 @> WARNING failed to parse beta-factor at line 9479 @> WARNING failed to parse beta-factor at line 9480 @> WARNING failed to parse beta-factor at line 9481 @> WARNING failed to parse beta-factor at line 9482 @> WARNING failed to parse beta-factor at line 9483 @> WARNING failed to parse beta-factor at line 9484 @> WARNING failed to parse beta-factor at line 9485 @> WARNING failed to parse beta-factor at line 9486 @> WARNING failed to parse beta-factor at line 9487 @> WARNING failed to parse beta-factor at line 9488 @> WARNING failed to parse beta-factor at line 9489 @> WARNING failed to parse beta-factor at line 9490 @> WARNING failed to parse beta-factor at line 9491 @> WARNING failed to parse beta-factor at line 9492 @> WARNING failed to parse beta-factor at line 9493 @> WARNING failed to parse beta-factor at line 9494 @> WARNING failed to parse beta-factor at line 9495 @> WARNING failed to parse beta-factor at line 9496 @> WARNING failed to parse beta-factor at line 9497 @> WARNING failed to parse beta-factor at line 9498 @> WARNING failed to parse beta-factor at line 9499 @> WARNING failed to parse beta-factor at line 9500 @> WARNING failed to parse beta-factor at line 9501 @> WARNING failed to parse beta-factor at line 9502 @> WARNING failed to parse beta-factor at line 9503 @> WARNING failed to parse beta-factor at line 9504 @> WARNING failed to parse beta-factor at line 9505 @> WARNING failed to parse beta-factor at line 9506 @> WARNING failed to parse beta-factor at line 9507 @> WARNING failed to parse beta-factor at line 9508 @> WARNING failed to parse beta-factor at line 9509 @> WARNING failed to parse beta-factor at line 9510 @> WARNING failed to parse beta-factor at line 9511 @> WARNING failed to parse beta-factor at line 9512 @> WARNING failed to parse beta-factor at line 9513 @> WARNING failed to parse beta-factor at line 9514 @> WARNING failed to parse beta-factor at line 9515 @> WARNING failed to parse beta-factor at line 9516 @> WARNING failed to parse beta-factor at line 9517 @> WARNING failed to parse beta-factor at line 9518 @> WARNING failed to parse beta-factor at line 9519 @> WARNING failed to parse beta-factor at line 9520 @> WARNING failed to parse beta-factor at line 9521 @> WARNING failed to parse beta-factor at line 9522 @> WARNING failed to parse beta-factor at line 9523 @> WARNING failed to parse beta-factor at line 9524 @> WARNING failed to parse beta-factor at line 9525 @> WARNING failed to parse beta-factor at line 9526 @> WARNING failed to parse beta-factor at line 9527 @> WARNING failed to parse beta-factor at line 9528 @> WARNING failed to parse beta-factor at line 9529 @> WARNING failed to parse beta-factor at line 9530 @> WARNING failed to parse beta-factor at line 9531 @> WARNING failed to parse beta-factor at line 9532 @> WARNING failed to parse beta-factor at line 9533 @> WARNING failed to parse beta-factor at line 9534 @> WARNING failed to parse beta-factor at line 9535 @> WARNING failed to parse beta-factor at line 9536 @> WARNING failed to parse beta-factor at line 9537 @> WARNING failed to parse beta-factor at line 9538 @> WARNING failed to parse beta-factor at line 9539 @> WARNING failed to parse beta-factor at line 9540 @> WARNING failed to parse beta-factor at line 9541 @> WARNING failed to parse beta-factor at line 9542 @> WARNING failed to parse beta-factor at line 9543 @> WARNING failed to parse beta-factor at line 9544 @> WARNING failed to parse beta-factor at line 9545 @> WARNING failed to parse beta-factor at line 9546 @> WARNING failed to parse beta-factor at line 9547 @> WARNING failed to parse beta-factor at line 9548 @> WARNING failed to parse beta-factor at line 9549 @> WARNING failed to parse beta-factor at line 9550 @> WARNING failed to parse beta-factor at line 9551 @> WARNING failed to parse beta-factor at line 9552 @> WARNING failed to parse beta-factor at line 9553 @> WARNING failed to parse beta-factor at line 9554 @> WARNING failed to parse beta-factor at line 9555 @> WARNING failed to parse beta-factor at line 9556 @> WARNING failed to parse beta-factor at line 9557 @> WARNING failed to parse beta-factor at line 9558 @> WARNING failed to parse beta-factor at line 9559 @> WARNING failed to parse beta-factor at line 9560 @> WARNING failed to parse beta-factor at line 9561 @> WARNING failed to parse beta-factor at line 9562 @> WARNING failed to parse beta-factor at line 9563 @> WARNING failed to parse beta-factor at line 9564 @> WARNING failed to parse beta-factor at line 9565 @> WARNING failed to parse beta-factor at line 9566 @> WARNING failed to parse beta-factor at line 9567 @> WARNING failed to parse beta-factor at line 9568 @> WARNING failed to parse beta-factor at line 9569 @> WARNING failed to parse beta-factor at line 9570 @> WARNING failed to parse beta-factor at line 9571 @> WARNING failed to parse beta-factor at line 9572 @> WARNING failed to parse beta-factor at line 9573 @> WARNING failed to parse beta-factor at line 9574 @> WARNING failed to parse beta-factor at line 9575 @> WARNING failed to parse beta-factor at line 9576 @> WARNING failed to parse beta-factor at line 9577 @> WARNING failed to parse beta-factor at line 9578 @> WARNING failed to parse beta-factor at line 9579 @> WARNING failed to parse beta-factor at line 9580 @> WARNING failed to parse beta-factor at line 9581 @> WARNING failed to parse beta-factor at line 9582 @> WARNING failed to parse beta-factor at line 9583 @> WARNING failed to parse beta-factor at line 9584 @> WARNING failed to parse beta-factor at line 9585 @> WARNING failed to parse beta-factor at line 9586 @> WARNING failed to parse beta-factor at line 9587 @> WARNING failed to parse beta-factor at line 9588 @> WARNING failed to parse beta-factor at line 9589 @> WARNING failed to parse beta-factor at line 9590 @> WARNING failed to parse beta-factor at line 9591 @> WARNING failed to parse beta-factor at line 9592 @> WARNING failed to parse beta-factor at line 9593 @> WARNING failed to parse beta-factor at line 9594 @> WARNING failed to parse beta-factor at line 9595 @> WARNING failed to parse beta-factor at line 9596 @> WARNING failed to parse beta-factor at line 9597 @> WARNING failed to parse beta-factor at line 9598 @> WARNING failed to parse beta-factor at line 9599 @> WARNING failed to parse beta-factor at line 9600 @> WARNING failed to parse beta-factor at line 9601 @> WARNING failed to parse beta-factor at line 9602 @> WARNING failed to parse beta-factor at line 9603 @> WARNING failed to parse beta-factor at line 9604 @> WARNING failed to parse beta-factor at line 9605 @> WARNING failed to parse beta-factor at line 9606 @> WARNING failed to parse beta-factor at line 9607 @> WARNING failed to parse beta-factor at line 9608 @> WARNING failed to parse beta-factor at line 9609 @> WARNING failed to parse beta-factor at line 9610 @> WARNING failed to parse beta-factor at line 9611 @> WARNING failed to parse beta-factor at line 9612 @> WARNING failed to parse beta-factor at line 9613 @> WARNING failed to parse beta-factor at line 9614 @> WARNING failed to parse beta-factor at line 9615 @> WARNING failed to parse beta-factor at line 9616 @> WARNING failed to parse beta-factor at line 9617 @> WARNING failed to parse beta-factor at line 9618 @> WARNING failed to parse beta-factor at line 9619 @> WARNING failed to parse beta-factor at line 9620 @> WARNING failed to parse beta-factor at line 9621 @> WARNING failed to parse beta-factor at line 9622 @> WARNING failed to parse beta-factor at line 9623 @> WARNING failed to parse beta-factor at line 9624 @> WARNING failed to parse beta-factor at line 9625 @> WARNING failed to parse beta-factor at line 9626 @> WARNING failed to parse beta-factor at line 9627 @> WARNING failed to parse beta-factor at line 9628 @> WARNING failed to parse beta-factor at line 9629 @> WARNING failed to parse beta-factor at line 9630 @> WARNING failed to parse beta-factor at line 9631 @> WARNING failed to parse beta-factor at line 9632 @> WARNING failed to parse beta-factor at line 9633 @> WARNING failed to parse beta-factor at line 9634 @> WARNING failed to parse beta-factor at line 9635 @> WARNING failed to parse beta-factor at line 9636 @> WARNING failed to parse beta-factor at line 9637 @> WARNING failed to parse beta-factor at line 9638 @> WARNING failed to parse beta-factor at line 9639 @> WARNING failed to parse beta-factor at line 9640 @> WARNING failed to parse beta-factor at line 9641 @> WARNING failed to parse beta-factor at line 9642 @> WARNING failed to parse beta-factor at line 9643 @> WARNING failed to parse beta-factor at line 9644 @> WARNING failed to parse beta-factor at line 9645 @> WARNING failed to parse beta-factor at line 9646 @> WARNING failed to parse beta-factor at line 9647 @> WARNING failed to parse beta-factor at line 9648 @> WARNING failed to parse beta-factor at line 9649 @> WARNING failed to parse beta-factor at line 9650 @> WARNING failed to parse beta-factor at line 9651 @> WARNING failed to parse beta-factor at line 9652 @> WARNING failed to parse beta-factor at line 9653 @> WARNING failed to parse beta-factor at line 9654 @> WARNING failed to parse beta-factor at line 9655 @> WARNING failed to parse beta-factor at line 9656 @> WARNING failed to parse beta-factor at line 9657 @> WARNING failed to parse beta-factor at line 9658 @> WARNING failed to parse beta-factor at line 9659 @> WARNING failed to parse beta-factor at line 9660 @> WARNING failed to parse beta-factor at line 9661 @> WARNING failed to parse beta-factor at line 9662 @> WARNING failed to parse beta-factor at line 9663 @> WARNING failed to parse beta-factor at line 9664 @> WARNING failed to parse beta-factor at line 9665 @> WARNING failed to parse beta-factor at line 9666 @> WARNING failed to parse beta-factor at line 9667 @> WARNING failed to parse beta-factor at line 9668 @> WARNING failed to parse beta-factor at line 9669 @> WARNING failed to parse beta-factor at line 9670 @> WARNING failed to parse beta-factor at line 9671 @> WARNING failed to parse beta-factor at line 9672 @> WARNING failed to parse beta-factor at line 9673 @> WARNING failed to parse beta-factor at line 9674 @> WARNING failed to parse beta-factor at line 9675 @> WARNING failed to parse beta-factor at line 9676 @> WARNING failed to parse beta-factor at line 9677 @> WARNING failed to parse beta-factor at line 9678 @> WARNING failed to parse beta-factor at line 9679 @> WARNING failed to parse beta-factor at line 9680 @> WARNING failed to parse beta-factor at line 9681 @> WARNING failed to parse beta-factor at line 9682 @> WARNING failed to parse beta-factor at line 9683 @> WARNING failed to parse beta-factor at line 9684 @> WARNING failed to parse beta-factor at line 9685 @> WARNING failed to parse beta-factor at line 9686 @> WARNING failed to parse beta-factor at line 9687 @> WARNING failed to parse beta-factor at line 9688 @> WARNING failed to parse beta-factor at line 9689 @> WARNING failed to parse beta-factor at line 9690 @> WARNING failed to parse beta-factor at line 9691 @> WARNING failed to parse beta-factor at line 9692 @> WARNING failed to parse beta-factor at line 9693 @> WARNING failed to parse beta-factor at line 9694 @> WARNING failed to parse beta-factor at line 9695 @> WARNING failed to parse beta-factor at line 9696 @> WARNING failed to parse beta-factor at line 9697 @> WARNING failed to parse beta-factor at line 9698 @> WARNING failed to parse beta-factor at line 9699 @> WARNING failed to parse beta-factor at line 9700 @> WARNING failed to parse beta-factor at line 9701 @> WARNING failed to parse beta-factor at line 9702 @> WARNING failed to parse beta-factor at line 9703 @> WARNING failed to parse beta-factor at line 9704 @> WARNING failed to parse beta-factor at line 9705 @> WARNING failed to parse beta-factor at line 9706 @> WARNING failed to parse beta-factor at line 9707 @> WARNING failed to parse beta-factor at line 9708 @> WARNING failed to parse beta-factor at line 9709 @> WARNING failed to parse beta-factor at line 9710 @> WARNING failed to parse beta-factor at line 9711 @> WARNING failed to parse beta-factor at line 9712 @> WARNING failed to parse beta-factor at line 9713 @> WARNING failed to parse beta-factor at line 9714 @> WARNING failed to parse beta-factor at line 9715 @> WARNING failed to parse beta-factor at line 9716 @> WARNING failed to parse beta-factor at line 9717 @> WARNING failed to parse beta-factor at line 9718 @> WARNING failed to parse beta-factor at line 9719 @> WARNING failed to parse beta-factor at line 9720 @> WARNING failed to parse beta-factor at line 9721 @> WARNING failed to parse beta-factor at line 9722 @> WARNING failed to parse beta-factor at line 9723 @> WARNING failed to parse beta-factor at line 9724 @> WARNING failed to parse beta-factor at line 9725 @> WARNING failed to parse beta-factor at line 9726 @> WARNING failed to parse beta-factor at line 9727 @> WARNING failed to parse beta-factor at line 9728 @> WARNING failed to parse beta-factor at line 9729 @> WARNING failed to parse beta-factor at line 9730 @> WARNING failed to parse beta-factor at line 9731 @> WARNING failed to parse beta-factor at line 9732 @> WARNING failed to parse beta-factor at line 9733 @> WARNING failed to parse beta-factor at line 9734 @> WARNING failed to parse beta-factor at line 9735 @> WARNING failed to parse beta-factor at line 9736 @> WARNING failed to parse beta-factor at line 9737 @> WARNING failed to parse beta-factor at line 9738 @> WARNING failed to parse beta-factor at line 9739 @> WARNING failed to parse beta-factor at line 9740 @> WARNING failed to parse beta-factor at line 9741 @> WARNING failed to parse beta-factor at line 9742 @> WARNING failed to parse beta-factor at line 9743 @> WARNING failed to parse beta-factor at line 9744 @> WARNING failed to parse beta-factor at line 9745 @> WARNING failed to parse beta-factor at line 9746 @> WARNING failed to parse beta-factor at line 9747 @> WARNING failed to parse beta-factor at line 9748 @> WARNING failed to parse beta-factor at line 9749 @> WARNING failed to parse beta-factor at line 9750 @> WARNING failed to parse beta-factor at line 9751 @> WARNING failed to parse beta-factor at line 9752 @> WARNING failed to parse beta-factor at line 9753 @> WARNING failed to parse beta-factor at line 9754 @> WARNING failed to parse beta-factor at line 9755 @> WARNING failed to parse beta-factor at line 9756 @> WARNING failed to parse beta-factor at line 9757 @> WARNING failed to parse beta-factor at line 9758 @> WARNING failed to parse beta-factor at line 9759 @> WARNING failed to parse beta-factor at line 9760 @> WARNING failed to parse beta-factor at line 9761 @> WARNING failed to parse beta-factor at line 9762 @> WARNING failed to parse beta-factor at line 9763 @> WARNING failed to parse beta-factor at line 9764 @> WARNING failed to parse beta-factor at line 9765 @> WARNING failed to parse beta-factor at line 9766 @> WARNING failed to parse beta-factor at line 9767 @> WARNING failed to parse beta-factor at line 9768 @> WARNING failed to parse beta-factor at line 9769 @> WARNING failed to parse beta-factor at line 9770 @> WARNING failed to parse beta-factor at line 9771 @> WARNING failed to parse beta-factor at line 9772 @> WARNING failed to parse beta-factor at line 9773 @> WARNING failed to parse beta-factor at line 9774 @> WARNING failed to parse beta-factor at line 9775 @> WARNING failed to parse beta-factor at line 9776 @> WARNING failed to parse beta-factor at line 9777 @> WARNING failed to parse beta-factor at line 9778 @> WARNING failed to parse beta-factor at line 9779 @> WARNING failed to parse beta-factor at line 9780 @> WARNING failed to parse beta-factor at line 9781 @> WARNING failed to parse beta-factor at line 9782 @> WARNING failed to parse beta-factor at line 9783 @> WARNING failed to parse beta-factor at line 9784 @> WARNING failed to parse beta-factor at line 9785 @> WARNING failed to parse beta-factor at line 9786 @> WARNING failed to parse beta-factor at line 9787 @> WARNING failed to parse beta-factor at line 9788 @> WARNING failed to parse beta-factor at line 9789 @> WARNING failed to parse beta-factor at line 9790 @> WARNING failed to parse beta-factor at line 9791 @> WARNING failed to parse beta-factor at line 9792 @> WARNING failed to parse beta-factor at line 9793 @> WARNING failed to parse beta-factor at line 9794 @> WARNING failed to parse beta-factor at line 9795 @> WARNING failed to parse beta-factor at line 9796 @> WARNING failed to parse beta-factor at line 9797 @> WARNING failed to parse beta-factor at line 9798 @> WARNING failed to parse beta-factor at line 9799 @> WARNING failed to parse beta-factor at line 9800 @> WARNING failed to parse beta-factor at line 9801 @> WARNING failed to parse beta-factor at line 9802 @> WARNING failed to parse beta-factor at line 9803 @> WARNING failed to parse beta-factor at line 9804 @> WARNING failed to parse beta-factor at line 9805 @> WARNING failed to parse beta-factor at line 9806 @> WARNING failed to parse beta-factor at line 9807 @> WARNING failed to parse beta-factor at line 9808 @> WARNING failed to parse beta-factor at line 9809 @> WARNING failed to parse beta-factor at line 9810 @> WARNING failed to parse beta-factor at line 9811 @> WARNING failed to parse beta-factor at line 9812 @> WARNING failed to parse beta-factor at line 9813 @> WARNING failed to parse beta-factor at line 9814 @> WARNING failed to parse beta-factor at line 9815 @> WARNING failed to parse beta-factor at line 9816 @> WARNING failed to parse beta-factor at line 9817 @> WARNING failed to parse beta-factor at line 9818 @> WARNING failed to parse beta-factor at line 9819 @> WARNING failed to parse beta-factor at line 9820 @> WARNING failed to parse beta-factor at line 9821 @> WARNING failed to parse beta-factor at line 9822 @> WARNING failed to parse beta-factor at line 9823 @> WARNING failed to parse beta-factor at line 9824 @> WARNING failed to parse beta-factor at line 9825 @> WARNING failed to parse beta-factor at line 9826 @> WARNING failed to parse beta-factor at line 9827 @> WARNING failed to parse beta-factor at line 9828 @> WARNING failed to parse beta-factor at line 9829 @> WARNING failed to parse beta-factor at line 9830 @> WARNING failed to parse beta-factor at line 9831 @> WARNING failed to parse beta-factor at line 9832 @> WARNING failed to parse beta-factor at line 9833 @> WARNING failed to parse beta-factor at line 9834 @> WARNING failed to parse beta-factor at line 9835 @> WARNING failed to parse beta-factor at line 9836 @> WARNING failed to parse beta-factor at line 9837 @> WARNING failed to parse beta-factor at line 9838 @> WARNING failed to parse beta-factor at line 9839 @> WARNING failed to parse beta-factor at line 9840 @> WARNING failed to parse beta-factor at line 9841 @> WARNING failed to parse beta-factor at line 9842 @> WARNING failed to parse beta-factor at line 9843 @> WARNING failed to parse beta-factor at line 9844 @> WARNING failed to parse beta-factor at line 9845 @> WARNING failed to parse beta-factor at line 9846 @> WARNING failed to parse beta-factor at line 9847 @> WARNING failed to parse beta-factor at line 9848 @> WARNING failed to parse beta-factor at line 9849 @> WARNING failed to parse beta-factor at line 9850 @> WARNING failed to parse beta-factor at line 9851 @> WARNING failed to parse beta-factor at line 9852 @> WARNING failed to parse beta-factor at line 9853 @> WARNING failed to parse beta-factor at line 9854 @> WARNING failed to parse beta-factor at line 9855 @> WARNING failed to parse beta-factor at line 9856 @> WARNING failed to parse beta-factor at line 9857 @> WARNING failed to parse beta-factor at line 9858 @> WARNING failed to parse beta-factor at line 9859 @> WARNING failed to parse beta-factor at line 9860 @> WARNING failed to parse beta-factor at line 9861 @> WARNING failed to parse beta-factor at line 9862 @> WARNING failed to parse beta-factor at line 9863 @> WARNING failed to parse beta-factor at line 9864 @> WARNING failed to parse beta-factor at line 9865 @> WARNING failed to parse beta-factor at line 9866 @> WARNING failed to parse beta-factor at line 9867 @> WARNING failed to parse beta-factor at line 9868 @> WARNING failed to parse beta-factor at line 9869 @> WARNING failed to parse beta-factor at line 9870 @> WARNING failed to parse beta-factor at line 9871 @> WARNING failed to parse beta-factor at line 9872 @> WARNING failed to parse beta-factor at line 9873 @> WARNING failed to parse beta-factor at line 9874 @> WARNING failed to parse beta-factor at line 9875 @> WARNING failed to parse beta-factor at line 9876 @> WARNING failed to parse beta-factor at line 9877 @> WARNING failed to parse beta-factor at line 9878 @> WARNING failed to parse beta-factor at line 9879 @> WARNING failed to parse beta-factor at line 9880 @> WARNING failed to parse beta-factor at line 9881 @> WARNING failed to parse beta-factor at line 9882 @> WARNING failed to parse beta-factor at line 9883 @> WARNING failed to parse beta-factor at line 9884 @> WARNING failed to parse beta-factor at line 9885 @> WARNING failed to parse beta-factor at line 9886 @> WARNING failed to parse beta-factor at line 9887 @> WARNING failed to parse beta-factor at line 9888 @> WARNING failed to parse beta-factor at line 9889 @> WARNING failed to parse beta-factor at line 9890 @> WARNING failed to parse beta-factor at line 9891 @> WARNING failed to parse beta-factor at line 9892 @> WARNING failed to parse beta-factor at line 9893 @> WARNING failed to parse beta-factor at line 9894 @> WARNING failed to parse beta-factor at line 9895 @> WARNING failed to parse beta-factor at line 9896 @> WARNING failed to parse beta-factor at line 9897 @> WARNING failed to parse beta-factor at line 9898 @> WARNING failed to parse beta-factor at line 9899 @> WARNING failed to parse beta-factor at line 9900 @> WARNING failed to parse beta-factor at line 9901 @> WARNING failed to parse beta-factor at line 9902 @> WARNING failed to parse beta-factor at line 9903 @> WARNING failed to parse beta-factor at line 9904 @> WARNING failed to parse beta-factor at line 9905 @> WARNING failed to parse beta-factor at line 9906 @> WARNING failed to parse beta-factor at line 9907 @> WARNING failed to parse beta-factor at line 9908 @> WARNING failed to parse beta-factor at line 9909 @> WARNING failed to parse beta-factor at line 9910 @> WARNING failed to parse beta-factor at line 9911 @> WARNING failed to parse beta-factor at line 9912 @> WARNING failed to parse beta-factor at line 9913 @> WARNING failed to parse beta-factor at line 9914 @> WARNING failed to parse beta-factor at line 9915 @> WARNING failed to parse beta-factor at line 9916 @> WARNING failed to parse beta-factor at line 9917 @> WARNING failed to parse beta-factor at line 9918 @> WARNING failed to parse beta-factor at line 9919 @> WARNING failed to parse beta-factor at line 9920 @> WARNING failed to parse beta-factor at line 9921 @> WARNING failed to parse beta-factor at line 9922 @> WARNING failed to parse beta-factor at line 9923 @> WARNING failed to parse beta-factor at line 9924 @> WARNING failed to parse beta-factor at line 9925 @> WARNING failed to parse beta-factor at line 9926 @> WARNING failed to parse beta-factor at line 9927 @> WARNING failed to parse beta-factor at line 9928 @> WARNING failed to parse beta-factor at line 9929 @> WARNING failed to parse beta-factor at line 9930 @> WARNING failed to parse beta-factor at line 9931 @> WARNING failed to parse beta-factor at line 9932 @> WARNING failed to parse beta-factor at line 9933 @> WARNING failed to parse beta-factor at line 9934 @> WARNING failed to parse beta-factor at line 9935 @> WARNING failed to parse beta-factor at line 9936 @> WARNING failed to parse beta-factor at line 9937 @> WARNING failed to parse beta-factor at line 9938 @> WARNING failed to parse beta-factor at line 9939 @> WARNING failed to parse beta-factor at line 9940 @> WARNING failed to parse beta-factor at line 9941 @> WARNING failed to parse beta-factor at line 9942 @> WARNING failed to parse beta-factor at line 9943 @> WARNING failed to parse beta-factor at line 9944 @> WARNING failed to parse beta-factor at line 9945 @> WARNING failed to parse beta-factor at line 9946 @> WARNING failed to parse beta-factor at line 9947 @> WARNING failed to parse beta-factor at line 9948 @> WARNING failed to parse beta-factor at line 9949 @> WARNING failed to parse beta-factor at line 9950 @> WARNING failed to parse beta-factor at line 9951 @> WARNING failed to parse beta-factor at line 9952 @> WARNING failed to parse beta-factor at line 9953 @> WARNING failed to parse beta-factor at line 9954 @> WARNING failed to parse beta-factor at line 9955 @> WARNING failed to parse beta-factor at line 9956 @> WARNING failed to parse beta-factor at line 9957 @> WARNING failed to parse beta-factor at line 9958 @> WARNING failed to parse beta-factor at line 9959 @> WARNING failed to parse beta-factor at line 9960 @> WARNING failed to parse beta-factor at line 9961 @> WARNING failed to parse beta-factor at line 9962 @> WARNING failed to parse beta-factor at line 9963 @> WARNING failed to parse beta-factor at line 9964 @> WARNING failed to parse beta-factor at line 9965 @> WARNING failed to parse beta-factor at line 9966 @> WARNING failed to parse beta-factor at line 9967 @> WARNING failed to parse beta-factor at line 9968 @> WARNING failed to parse beta-factor at line 9969 @> WARNING failed to parse beta-factor at line 9970 @> WARNING failed to parse beta-factor at line 9971 @> WARNING failed to parse beta-factor at line 9972 @> WARNING failed to parse beta-factor at line 9973 @> WARNING failed to parse beta-factor at line 9974 @> WARNING failed to parse beta-factor at line 9975 @> WARNING failed to parse beta-factor at line 9976 @> WARNING failed to parse beta-factor at line 9977 @> WARNING failed to parse beta-factor at line 9978 @> WARNING failed to parse beta-factor at line 9979 @> WARNING failed to parse beta-factor at line 9980 @> WARNING failed to parse beta-factor at line 9981 @> WARNING failed to parse beta-factor at line 9982 @> WARNING failed to parse beta-factor at line 9983 @> WARNING failed to parse beta-factor at line 9984 @> WARNING failed to parse beta-factor at line 9985 @> WARNING failed to parse beta-factor at line 9986 @> WARNING failed to parse beta-factor at line 9987 @> WARNING failed to parse beta-factor at line 9988 @> WARNING failed to parse beta-factor at line 9989 @> WARNING failed to parse beta-factor at line 9990 @> WARNING failed to parse beta-factor at line 9991 @> WARNING failed to parse beta-factor at line 9992 @> WARNING failed to parse beta-factor at line 9993 @> WARNING failed to parse beta-factor at line 9994 @> WARNING failed to parse beta-factor at line 9995 @> WARNING failed to parse beta-factor at line 9996 @> WARNING failed to parse beta-factor at line 9997 @> WARNING failed to parse beta-factor at line 9998 @> WARNING failed to parse beta-factor at line 9999 @> WARNING failed to parse beta-factor at line 10000 @> WARNING failed to parse beta-factor at line 10001 @> WARNING failed to parse beta-factor at line 10002 @> WARNING failed to parse beta-factor at line 10003 @> WARNING failed to parse beta-factor at line 10004 @> WARNING failed to parse beta-factor at line 10005 @> WARNING failed to parse beta-factor at line 10006 @> WARNING failed to parse beta-factor at line 10007 @> WARNING failed to parse beta-factor at line 10008 @> WARNING failed to parse beta-factor at line 10009 @> WARNING failed to parse beta-factor at line 10010 @> WARNING failed to parse beta-factor at line 10011 @> WARNING failed to parse beta-factor at line 10012 @> WARNING failed to parse beta-factor at line 10013 @> WARNING failed to parse beta-factor at line 10014 @> WARNING failed to parse beta-factor at line 10015 @> WARNING failed to parse beta-factor at line 10016 @> WARNING failed to parse beta-factor at line 10017 @> WARNING failed to parse beta-factor at line 10018 @> WARNING failed to parse beta-factor at line 10019 @> WARNING failed to parse beta-factor at line 10020 @> WARNING failed to parse beta-factor at line 10021 @> WARNING failed to parse beta-factor at line 10022 @> WARNING failed to parse beta-factor at line 10023 @> WARNING failed to parse beta-factor at line 10024 @> WARNING failed to parse beta-factor at line 10025 @> WARNING failed to parse beta-factor at line 10026 @> WARNING failed to parse beta-factor at line 10027 @> WARNING failed to parse beta-factor at line 10028 @> WARNING failed to parse beta-factor at line 10029 @> WARNING failed to parse beta-factor at line 10030 @> WARNING failed to parse beta-factor at line 10031 @> WARNING failed to parse beta-factor at line 10032 @> WARNING failed to parse beta-factor at line 10033 @> WARNING failed to parse beta-factor at line 10034 @> WARNING failed to parse beta-factor at line 10035 @> WARNING failed to parse beta-factor at line 10036 @> WARNING failed to parse beta-factor at line 10037 @> WARNING failed to parse beta-factor at line 10038 @> WARNING failed to parse beta-factor at line 10039 @> WARNING failed to parse beta-factor at line 10040 @> WARNING failed to parse beta-factor at line 10041 @> WARNING failed to parse beta-factor at line 10042 @> WARNING failed to parse beta-factor at line 10043 @> WARNING failed to parse beta-factor at line 10044 @> WARNING failed to parse beta-factor at line 10045 @> WARNING failed to parse beta-factor at line 10046 @> WARNING failed to parse beta-factor at line 10047 @> WARNING failed to parse beta-factor at line 10048 @> WARNING failed to parse beta-factor at line 10049 @> WARNING failed to parse beta-factor at line 10050 @> WARNING failed to parse beta-factor at line 10051 @> WARNING failed to parse beta-factor at line 10052 @> WARNING failed to parse beta-factor at line 10053 @> WARNING failed to parse beta-factor at line 10054 @> WARNING failed to parse beta-factor at line 10055 @> WARNING failed to parse beta-factor at line 10056 @> WARNING failed to parse beta-factor at line 10057 @> WARNING failed to parse beta-factor at line 10058 @> WARNING failed to parse beta-factor at line 10059 @> WARNING failed to parse beta-factor at line 10060 @> WARNING failed to parse beta-factor at line 10061 @> WARNING failed to parse beta-factor at line 10062 @> WARNING failed to parse beta-factor at line 10063 @> WARNING failed to parse beta-factor at line 10064 @> WARNING failed to parse beta-factor at line 10065 @> WARNING failed to parse beta-factor at line 10066 @> WARNING failed to parse beta-factor at line 10067 @> WARNING failed to parse beta-factor at line 10068 @> WARNING failed to parse beta-factor at line 10069 @> WARNING failed to parse beta-factor at line 10070 @> WARNING failed to parse beta-factor at line 10071 @> WARNING failed to parse beta-factor at line 10072 @> WARNING failed to parse beta-factor at line 10073 @> WARNING failed to parse beta-factor at line 10074 @> WARNING failed to parse beta-factor at line 10075 @> WARNING failed to parse beta-factor at line 10076 @> WARNING failed to parse beta-factor at line 10077 @> WARNING failed to parse beta-factor at line 10078 @> WARNING failed to parse beta-factor at line 10079 @> WARNING failed to parse beta-factor at line 10080 @> WARNING failed to parse beta-factor at line 10081 @> WARNING failed to parse beta-factor at line 10082 @> WARNING failed to parse beta-factor at line 10083 @> WARNING failed to parse beta-factor at line 10084 @> WARNING failed to parse beta-factor at line 10085 @> WARNING failed to parse beta-factor at line 10086 @> WARNING failed to parse beta-factor at line 10087 @> WARNING failed to parse beta-factor at line 10088 @> WARNING failed to parse beta-factor at line 10089 @> WARNING failed to parse beta-factor at line 10090 @> WARNING failed to parse beta-factor at line 10091 @> WARNING failed to parse beta-factor at line 10092 @> WARNING failed to parse beta-factor at line 10093 @> WARNING failed to parse beta-factor at line 10094 @> WARNING failed to parse beta-factor at line 10095 @> WARNING failed to parse beta-factor at line 10096 @> WARNING failed to parse beta-factor at line 10097 @> WARNING failed to parse beta-factor at line 10098 @> WARNING failed to parse beta-factor at line 10099 @> WARNING failed to parse beta-factor at line 10100 @> WARNING failed to parse beta-factor at line 10101 @> WARNING failed to parse beta-factor at line 10102 @> WARNING failed to parse beta-factor at line 10103 @> WARNING failed to parse beta-factor at line 10104 @> WARNING failed to parse beta-factor at line 10105 @> WARNING failed to parse beta-factor at line 10106 @> WARNING failed to parse beta-factor at line 10107 @> WARNING failed to parse beta-factor at line 10108 @> WARNING failed to parse beta-factor at line 10109 @> WARNING failed to parse beta-factor at line 10110 @> WARNING failed to parse beta-factor at line 10111 @> WARNING failed to parse beta-factor at line 10112 @> WARNING failed to parse beta-factor at line 10113 @> WARNING failed to parse beta-factor at line 10114 @> WARNING failed to parse beta-factor at line 10115 @> WARNING failed to parse beta-factor at line 10116 @> WARNING failed to parse beta-factor at line 10117 @> WARNING failed to parse beta-factor at line 10118 @> WARNING failed to parse beta-factor at line 10119 @> WARNING failed to parse beta-factor at line 10120 @> WARNING failed to parse beta-factor at line 10121 @> WARNING failed to parse beta-factor at line 10122 @> WARNING failed to parse beta-factor at line 10123 @> WARNING failed to parse beta-factor at line 10124 @> WARNING failed to parse beta-factor at line 10125 @> WARNING failed to parse beta-factor at line 10126 @> WARNING failed to parse beta-factor at line 10127 @> WARNING failed to parse beta-factor at line 10128 @> WARNING failed to parse beta-factor at line 10129 @> WARNING failed to parse beta-factor at line 10130 @> WARNING failed to parse beta-factor at line 10131 @> WARNING failed to parse beta-factor at line 10132 @> WARNING failed to parse beta-factor at line 10133 @> WARNING failed to parse beta-factor at line 10134 @> WARNING failed to parse beta-factor at line 10135 @> WARNING failed to parse beta-factor at line 10136 @> WARNING failed to parse beta-factor at line 10137 @> WARNING failed to parse beta-factor at line 10138 @> WARNING failed to parse beta-factor at line 10139 @> WARNING failed to parse beta-factor at line 10140 @> WARNING failed to parse beta-factor at line 10141 @> WARNING failed to parse beta-factor at line 10142 @> WARNING failed to parse beta-factor at line 10143 @> WARNING failed to parse beta-factor at line 10144 @> WARNING failed to parse beta-factor at line 10145 @> WARNING failed to parse beta-factor at line 10146 @> WARNING failed to parse beta-factor at line 10147 @> WARNING failed to parse beta-factor at line 10148 @> WARNING failed to parse beta-factor at line 10149 @> WARNING failed to parse beta-factor at line 10150 @> WARNING failed to parse beta-factor at line 10151 @> WARNING failed to parse beta-factor at line 10152 @> WARNING failed to parse beta-factor at line 10153 @> WARNING failed to parse beta-factor at line 10154 @> WARNING failed to parse beta-factor at line 10155 @> WARNING failed to parse beta-factor at line 10156 @> WARNING failed to parse beta-factor at line 10157 @> WARNING failed to parse beta-factor at line 10158 @> WARNING failed to parse beta-factor at line 10159 @> WARNING failed to parse beta-factor at line 10160 @> WARNING failed to parse beta-factor at line 10161 @> WARNING failed to parse beta-factor at line 10162 @> WARNING failed to parse beta-factor at line 10163 @> WARNING failed to parse beta-factor at line 10164 @> WARNING failed to parse beta-factor at line 10165 @> WARNING failed to parse beta-factor at line 10166 @> WARNING failed to parse beta-factor at line 10167 @> WARNING failed to parse beta-factor at line 10168 @> WARNING failed to parse beta-factor at line 10169 @> WARNING failed to parse beta-factor at line 10170 @> WARNING failed to parse beta-factor at line 10171 @> WARNING failed to parse beta-factor at line 10172 @> WARNING failed to parse beta-factor at line 10173 @> WARNING failed to parse beta-factor at line 10174 @> WARNING failed to parse beta-factor at line 10175 @> WARNING failed to parse beta-factor at line 10176 @> WARNING failed to parse beta-factor at line 10177 @> WARNING failed to parse beta-factor at line 10178 @> WARNING failed to parse beta-factor at line 10179 @> WARNING failed to parse beta-factor at line 10180 @> WARNING failed to parse beta-factor at line 10181 @> WARNING failed to parse beta-factor at line 10182 @> WARNING failed to parse beta-factor at line 10183 @> WARNING failed to parse beta-factor at line 10184 @> WARNING failed to parse beta-factor at line 10185 @> WARNING failed to parse beta-factor at line 10186 @> WARNING failed to parse beta-factor at line 10187 @> WARNING failed to parse beta-factor at line 10188 @> WARNING failed to parse beta-factor at line 10189 @> WARNING failed to parse beta-factor at line 10190 @> WARNING failed to parse beta-factor at line 10191 @> WARNING failed to parse beta-factor at line 10192 @> WARNING failed to parse beta-factor at line 10193 @> WARNING failed to parse beta-factor at line 10194 @> WARNING failed to parse beta-factor at line 10195 @> WARNING failed to parse beta-factor at line 10196 @> WARNING failed to parse beta-factor at line 10197 @> WARNING failed to parse beta-factor at line 10198 @> WARNING failed to parse beta-factor at line 10199 @> WARNING failed to parse beta-factor at line 10200 @> WARNING failed to parse beta-factor at line 10201 @> WARNING failed to parse beta-factor at line 10202 @> WARNING failed to parse beta-factor at line 10203 @> WARNING failed to parse beta-factor at line 10204 @> WARNING failed to parse beta-factor at line 10205 @> WARNING failed to parse beta-factor at line 10206 @> WARNING failed to parse beta-factor at line 10207 @> WARNING failed to parse beta-factor at line 10208 @> WARNING failed to parse beta-factor at line 10209 @> WARNING failed to parse beta-factor at line 10210 @> WARNING failed to parse beta-factor at line 10211 @> WARNING failed to parse beta-factor at line 10212 @> WARNING failed to parse beta-factor at line 10213 @> WARNING failed to parse beta-factor at line 10214 @> WARNING failed to parse beta-factor at line 10215 @> WARNING failed to parse beta-factor at line 10216 @> WARNING failed to parse beta-factor at line 10217 @> WARNING failed to parse beta-factor at line 10218 @> WARNING failed to parse beta-factor at line 10219 @> WARNING failed to parse beta-factor at line 10220 @> WARNING failed to parse beta-factor at line 10221 @> WARNING failed to parse beta-factor at line 10222 @> WARNING failed to parse beta-factor at line 10223 @> WARNING failed to parse beta-factor at line 10224 @> WARNING failed to parse beta-factor at line 10225 @> WARNING failed to parse beta-factor at line 10226 @> WARNING failed to parse beta-factor at line 10227 @> WARNING failed to parse beta-factor at line 10228 @> WARNING failed to parse beta-factor at line 10229 @> WARNING failed to parse beta-factor at line 10230 @> WARNING failed to parse beta-factor at line 10231 @> WARNING failed to parse beta-factor at line 10232 @> WARNING failed to parse beta-factor at line 10233 @> WARNING failed to parse beta-factor at line 10234 @> WARNING failed to parse beta-factor at line 10235 @> WARNING failed to parse beta-factor at line 10236 @> WARNING failed to parse beta-factor at line 10237 @> WARNING failed to parse beta-factor at line 10238 @> WARNING failed to parse beta-factor at line 10239 @> WARNING failed to parse beta-factor at line 10240 @> WARNING failed to parse beta-factor at line 10241 @> WARNING failed to parse beta-factor at line 10242 @> WARNING failed to parse beta-factor at line 10243 @> WARNING failed to parse beta-factor at line 10244 @> WARNING failed to parse beta-factor at line 10245 @> WARNING failed to parse beta-factor at line 10246 @> WARNING failed to parse beta-factor at line 10247 @> WARNING failed to parse beta-factor at line 10248 @> WARNING failed to parse beta-factor at line 10249 @> WARNING failed to parse beta-factor at line 10250 @> WARNING failed to parse beta-factor at line 10251 @> WARNING failed to parse beta-factor at line 10252 @> WARNING failed to parse beta-factor at line 10253 @> WARNING failed to parse beta-factor at line 10254 @> WARNING failed to parse beta-factor at line 10255 @> WARNING failed to parse beta-factor at line 10256 @> WARNING failed to parse beta-factor at line 10257 @> WARNING failed to parse beta-factor at line 10258 @> WARNING failed to parse beta-factor at line 10259 @> WARNING failed to parse beta-factor at line 10260 @> WARNING failed to parse beta-factor at line 10261 @> WARNING failed to parse beta-factor at line 10262 @> WARNING failed to parse beta-factor at line 10263 @> WARNING failed to parse beta-factor at line 10264 @> WARNING failed to parse beta-factor at line 10265 @> WARNING failed to parse beta-factor at line 10266 @> WARNING failed to parse beta-factor at line 10267 @> WARNING failed to parse beta-factor at line 10268 @> WARNING failed to parse beta-factor at line 10269 @> WARNING failed to parse beta-factor at line 10270 @> WARNING failed to parse beta-factor at line 10271 @> WARNING failed to parse beta-factor at line 10272 @> WARNING failed to parse beta-factor at line 10273 @> WARNING failed to parse beta-factor at line 10274 @> WARNING failed to parse beta-factor at line 10275 @> WARNING failed to parse beta-factor at line 10276 @> WARNING failed to parse beta-factor at line 10277 @> WARNING failed to parse beta-factor at line 10278 @> WARNING failed to parse beta-factor at line 10279 @> WARNING failed to parse beta-factor at line 10280 @> WARNING failed to parse beta-factor at line 10281 @> WARNING failed to parse beta-factor at line 10282 @> WARNING failed to parse beta-factor at line 10283 @> WARNING failed to parse beta-factor at line 10284 @> WARNING failed to parse beta-factor at line 10285 @> WARNING failed to parse beta-factor at line 10286 @> WARNING failed to parse beta-factor at line 10287 @> WARNING failed to parse beta-factor at line 10288 @> WARNING failed to parse beta-factor at line 10289 @> WARNING failed to parse beta-factor at line 10290 @> WARNING failed to parse beta-factor at line 10291 @> WARNING failed to parse beta-factor at line 10292 @> WARNING failed to parse beta-factor at line 10293 @> WARNING failed to parse beta-factor at line 10294 @> WARNING failed to parse beta-factor at line 10295 @> WARNING failed to parse beta-factor at line 10296 @> WARNING failed to parse beta-factor at line 10297 @> WARNING failed to parse beta-factor at line 10298 @> WARNING failed to parse beta-factor at line 10299 @> WARNING failed to parse beta-factor at line 10300 @> WARNING failed to parse beta-factor at line 10301 @> WARNING failed to parse beta-factor at line 10302 @> WARNING failed to parse beta-factor at line 10303 @> WARNING failed to parse beta-factor at line 10304 @> WARNING failed to parse beta-factor at line 10305 @> WARNING failed to parse beta-factor at line 10306 @> WARNING failed to parse beta-factor at line 10307 @> WARNING failed to parse beta-factor at line 10308 @> WARNING failed to parse beta-factor at line 10309 @> WARNING failed to parse beta-factor at line 10310 @> WARNING failed to parse beta-factor at line 10311 @> WARNING failed to parse beta-factor at line 10312 @> WARNING failed to parse beta-factor at line 10313 @> WARNING failed to parse beta-factor at line 10314 @> WARNING failed to parse beta-factor at line 10315 @> WARNING failed to parse beta-factor at line 10316 @> WARNING failed to parse beta-factor at line 10317 @> WARNING failed to parse beta-factor at line 10318 @> WARNING failed to parse beta-factor at line 10319 @> WARNING failed to parse beta-factor at line 10320 @> WARNING failed to parse beta-factor at line 10321 @> WARNING failed to parse beta-factor at line 10322 @> WARNING failed to parse beta-factor at line 10323 @> WARNING failed to parse beta-factor at line 10324 @> WARNING failed to parse beta-factor at line 10325 @> WARNING failed to parse beta-factor at line 10326 @> WARNING failed to parse beta-factor at line 10327 @> WARNING failed to parse beta-factor at line 10328 @> WARNING failed to parse beta-factor at line 10329 @> WARNING failed to parse beta-factor at line 10330 @> WARNING failed to parse beta-factor at line 10331 @> WARNING failed to parse beta-factor at line 10332 @> WARNING failed to parse beta-factor at line 10333 @> WARNING failed to parse beta-factor at line 10334 @> WARNING failed to parse beta-factor at line 10335 @> WARNING failed to parse beta-factor at line 10336 @> WARNING failed to parse beta-factor at line 10337 @> WARNING failed to parse beta-factor at line 10338 @> WARNING failed to parse beta-factor at line 10339 @> WARNING failed to parse beta-factor at line 10340 @> WARNING failed to parse beta-factor at line 10341 @> WARNING failed to parse beta-factor at line 10342 @> WARNING failed to parse beta-factor at line 10343 @> WARNING failed to parse beta-factor at line 10344 @> WARNING failed to parse beta-factor at line 10345 @> WARNING failed to parse beta-factor at line 10346 @> WARNING failed to parse beta-factor at line 10347 @> WARNING failed to parse beta-factor at line 10348 @> WARNING failed to parse beta-factor at line 10349 @> WARNING failed to parse beta-factor at line 10350 @> WARNING failed to parse beta-factor at line 10351 @> WARNING failed to parse beta-factor at line 10352 @> WARNING failed to parse beta-factor at line 10353 @> WARNING failed to parse beta-factor at line 10354 @> WARNING failed to parse beta-factor at line 10355 @> WARNING failed to parse beta-factor at line 10356 @> WARNING failed to parse beta-factor at line 10357 @> WARNING failed to parse beta-factor at line 10358 @> WARNING failed to parse beta-factor at line 10359 @> WARNING failed to parse beta-factor at line 10360 @> WARNING failed to parse beta-factor at line 10361 @> WARNING failed to parse beta-factor at line 10362 @> WARNING failed to parse beta-factor at line 10363 @> WARNING failed to parse beta-factor at line 10364 @> WARNING failed to parse beta-factor at line 10365 @> WARNING failed to parse beta-factor at line 10366 @> WARNING failed to parse beta-factor at line 10367 @> WARNING failed to parse beta-factor at line 10368 @> WARNING failed to parse beta-factor at line 10369 @> WARNING failed to parse beta-factor at line 10370 @> WARNING failed to parse beta-factor at line 10371 @> WARNING failed to parse beta-factor at line 10372 @> WARNING failed to parse beta-factor at line 10373 @> WARNING failed to parse beta-factor at line 10374 @> WARNING failed to parse beta-factor at line 10375 @> WARNING failed to parse beta-factor at line 10376 @> WARNING failed to parse beta-factor at line 10377 @> WARNING failed to parse beta-factor at line 10378 @> WARNING failed to parse beta-factor at line 10379 @> WARNING failed to parse beta-factor at line 10380 @> WARNING failed to parse beta-factor at line 10381 @> WARNING failed to parse beta-factor at line 10382 @> WARNING failed to parse beta-factor at line 10383 @> WARNING failed to parse beta-factor at line 10384 @> WARNING failed to parse beta-factor at line 10385 @> WARNING failed to parse beta-factor at line 10386 @> WARNING failed to parse beta-factor at line 10387 @> WARNING failed to parse beta-factor at line 10388 @> WARNING failed to parse beta-factor at line 10389 @> WARNING failed to parse beta-factor at line 10390 @> WARNING failed to parse beta-factor at line 10391 @> WARNING failed to parse beta-factor at line 10392 @> WARNING failed to parse beta-factor at line 10393 @> WARNING failed to parse beta-factor at line 10394 @> WARNING failed to parse beta-factor at line 10395 @> WARNING failed to parse beta-factor at line 10396 @> WARNING failed to parse beta-factor at line 10397 @> WARNING failed to parse beta-factor at line 10398 @> WARNING failed to parse beta-factor at line 10399 @> WARNING failed to parse beta-factor at line 10400 @> WARNING failed to parse beta-factor at line 10401 @> WARNING failed to parse beta-factor at line 10402 @> WARNING failed to parse beta-factor at line 10403 @> WARNING failed to parse beta-factor at line 10404 @> WARNING failed to parse beta-factor at line 10405 @> WARNING failed to parse beta-factor at line 10406 @> WARNING failed to parse beta-factor at line 10407 @> WARNING failed to parse beta-factor at line 10408 @> WARNING failed to parse beta-factor at line 10409 @> WARNING failed to parse beta-factor at line 10410 @> WARNING failed to parse beta-factor at line 10411 @> WARNING failed to parse beta-factor at line 10412 @> WARNING failed to parse beta-factor at line 10413 @> WARNING failed to parse beta-factor at line 10414 @> WARNING failed to parse beta-factor at line 10415 @> WARNING failed to parse beta-factor at line 10416 @> WARNING failed to parse beta-factor at line 10417 @> WARNING failed to parse beta-factor at line 10418 @> WARNING failed to parse beta-factor at line 10419 @> WARNING failed to parse beta-factor at line 10420 @> WARNING failed to parse beta-factor at line 10421 @> WARNING failed to parse beta-factor at line 10422 @> WARNING failed to parse beta-factor at line 10423 @> WARNING failed to parse beta-factor at line 10424 @> WARNING failed to parse beta-factor at line 10425 @> WARNING failed to parse beta-factor at line 10426 @> WARNING failed to parse beta-factor at line 10427 @> WARNING failed to parse beta-factor at line 10428 @> WARNING failed to parse beta-factor at line 10429 @> WARNING failed to parse beta-factor at line 10430 @> WARNING failed to parse beta-factor at line 10431 @> WARNING failed to parse beta-factor at line 10432 @> WARNING failed to parse beta-factor at line 10433 @> WARNING failed to parse beta-factor at line 10434 @> WARNING failed to parse beta-factor at line 10435 @> WARNING failed to parse beta-factor at line 10436 @> WARNING failed to parse beta-factor at line 10437 @> WARNING failed to parse beta-factor at line 10438 @> WARNING failed to parse beta-factor at line 10439 @> WARNING failed to parse beta-factor at line 10440 @> WARNING failed to parse beta-factor at line 10441 @> WARNING failed to parse beta-factor at line 10442 @> WARNING failed to parse beta-factor at line 10443 @> WARNING failed to parse beta-factor at line 10444 @> WARNING failed to parse beta-factor at line 10445 @> WARNING failed to parse beta-factor at line 10446 @> WARNING failed to parse beta-factor at line 10447 @> WARNING failed to parse beta-factor at line 10448 @> WARNING failed to parse beta-factor at line 10449 @> WARNING failed to parse beta-factor at line 10450 @> WARNING failed to parse beta-factor at line 10451 @> WARNING failed to parse beta-factor at line 10452 @> WARNING failed to parse beta-factor at line 10453 @> WARNING failed to parse beta-factor at line 10454 @> WARNING failed to parse beta-factor at line 10455 @> WARNING failed to parse beta-factor at line 10456 @> WARNING failed to parse beta-factor at line 10457 @> WARNING failed to parse beta-factor at line 10458 @> WARNING failed to parse beta-factor at line 10459 @> WARNING failed to parse beta-factor at line 10460 @> WARNING failed to parse beta-factor at line 10461 @> WARNING failed to parse beta-factor at line 10462 @> WARNING failed to parse beta-factor at line 10463 @> WARNING failed to parse beta-factor at line 10464 @> WARNING failed to parse beta-factor at line 10465 @> WARNING failed to parse beta-factor at line 10466 @> WARNING failed to parse beta-factor at line 10467 @> WARNING failed to parse beta-factor at line 10468 @> WARNING failed to parse beta-factor at line 10469 @> WARNING failed to parse beta-factor at line 10470 @> WARNING failed to parse beta-factor at line 10471 @> WARNING failed to parse beta-factor at line 10472 @> WARNING failed to parse beta-factor at line 10473 @> WARNING failed to parse beta-factor at line 10474 @> WARNING failed to parse beta-factor at line 10475 @> WARNING failed to parse beta-factor at line 10476 @> WARNING failed to parse beta-factor at line 10477 @> WARNING failed to parse beta-factor at line 10478 @> WARNING failed to parse beta-factor at line 10479 @> WARNING failed to parse beta-factor at line 10480 @> WARNING failed to parse beta-factor at line 10481 @> WARNING failed to parse beta-factor at line 10482 @> WARNING failed to parse beta-factor at line 10483 @> WARNING failed to parse beta-factor at line 10484 @> WARNING failed to parse beta-factor at line 10485 @> WARNING failed to parse beta-factor at line 10486 @> WARNING failed to parse beta-factor at line 10487 @> WARNING failed to parse beta-factor at line 10488 @> WARNING failed to parse beta-factor at line 10489 @> WARNING failed to parse beta-factor at line 10490 @> WARNING failed to parse beta-factor at line 10491 @> WARNING failed to parse beta-factor at line 10492 @> WARNING failed to parse beta-factor at line 10493 @> WARNING failed to parse beta-factor at line 10494 @> WARNING failed to parse beta-factor at line 10495 @> WARNING failed to parse beta-factor at line 10496 @> WARNING failed to parse beta-factor at line 10497 @> WARNING failed to parse beta-factor at line 10498 @> WARNING failed to parse beta-factor at line 10499 @> WARNING failed to parse beta-factor at line 10500 @> WARNING failed to parse beta-factor at line 10501 @> WARNING failed to parse beta-factor at line 10502 @> WARNING failed to parse beta-factor at line 10503 @> WARNING failed to parse beta-factor at line 10504 @> WARNING failed to parse beta-factor at line 10505 @> WARNING failed to parse beta-factor at line 10506 @> WARNING failed to parse beta-factor at line 10507 @> WARNING failed to parse beta-factor at line 10508 @> WARNING failed to parse beta-factor at line 10509 @> WARNING failed to parse beta-factor at line 10510 @> WARNING failed to parse beta-factor at line 10511 @> WARNING failed to parse beta-factor at line 10512 @> WARNING failed to parse beta-factor at line 10513 @> WARNING failed to parse beta-factor at line 10514 @> WARNING failed to parse beta-factor at line 10515 @> WARNING failed to parse beta-factor at line 10516 @> WARNING failed to parse beta-factor at line 10517 @> WARNING failed to parse beta-factor at line 10518 @> WARNING failed to parse beta-factor at line 10519 @> WARNING failed to parse beta-factor at line 10520 @> WARNING failed to parse beta-factor at line 10521 @> WARNING failed to parse beta-factor at line 10522 @> WARNING failed to parse beta-factor at line 10523 @> WARNING failed to parse beta-factor at line 10524 @> WARNING failed to parse beta-factor at line 10525 @> WARNING failed to parse beta-factor at line 10526 @> WARNING failed to parse beta-factor at line 10527 @> WARNING failed to parse beta-factor at line 10528 @> WARNING failed to parse beta-factor at line 10529 @> WARNING failed to parse beta-factor at line 10530 @> WARNING failed to parse beta-factor at line 10531 @> WARNING failed to parse beta-factor at line 10532 @> WARNING failed to parse beta-factor at line 10533 @> WARNING failed to parse beta-factor at line 10534 @> WARNING failed to parse beta-factor at line 10535 @> WARNING failed to parse beta-factor at line 10536 @> WARNING failed to parse beta-factor at line 10537 @> WARNING failed to parse beta-factor at line 10538 @> WARNING failed to parse beta-factor at line 10539 @> WARNING failed to parse beta-factor at line 10540 @> WARNING failed to parse beta-factor at line 10541 @> WARNING failed to parse beta-factor at line 10542 @> WARNING failed to parse beta-factor at line 10543 @> WARNING failed to parse beta-factor at line 10544 @> WARNING failed to parse beta-factor at line 10545 @> WARNING failed to parse beta-factor at line 10546 @> WARNING failed to parse beta-factor at line 10547 @> WARNING failed to parse beta-factor at line 10548 @> WARNING failed to parse beta-factor at line 10549 @> WARNING failed to parse beta-factor at line 10550 @> WARNING failed to parse beta-factor at line 10551 @> WARNING failed to parse beta-factor at line 10552 @> WARNING failed to parse beta-factor at line 10553 @> WARNING failed to parse beta-factor at line 10554 @> WARNING failed to parse beta-factor at line 10555 @> WARNING failed to parse beta-factor at line 10556 @> WARNING failed to parse beta-factor at line 10557 @> WARNING failed to parse beta-factor at line 10558 @> WARNING failed to parse beta-factor at line 10559 @> WARNING failed to parse beta-factor at line 10560 @> WARNING failed to parse beta-factor at line 10561 @> WARNING failed to parse beta-factor at line 10562 @> WARNING failed to parse beta-factor at line 10563 @> WARNING failed to parse beta-factor at line 10564 @> WARNING failed to parse beta-factor at line 10565 @> WARNING failed to parse beta-factor at line 10566 @> WARNING failed to parse beta-factor at line 10567 @> WARNING failed to parse beta-factor at line 10568 @> WARNING failed to parse beta-factor at line 10569 @> WARNING failed to parse beta-factor at line 10570 @> WARNING failed to parse beta-factor at line 10571 @> WARNING failed to parse beta-factor at line 10572 @> WARNING failed to parse beta-factor at line 10573 @> WARNING failed to parse beta-factor at line 10574 @> WARNING failed to parse beta-factor at line 10575 @> WARNING failed to parse beta-factor at line 10576 @> WARNING failed to parse beta-factor at line 10577 @> WARNING failed to parse beta-factor at line 10578 @> WARNING failed to parse beta-factor at line 10579 @> WARNING failed to parse beta-factor at line 10580 @> WARNING failed to parse beta-factor at line 10581 @> WARNING failed to parse beta-factor at line 10582 @> WARNING failed to parse beta-factor at line 10583 @> WARNING failed to parse beta-factor at line 10584 @> WARNING failed to parse beta-factor at line 10585 @> WARNING failed to parse beta-factor at line 10586 @> WARNING failed to parse beta-factor at line 10587 @> WARNING failed to parse beta-factor at line 10588 @> WARNING failed to parse beta-factor at line 10589 @> WARNING failed to parse beta-factor at line 10590 @> WARNING failed to parse beta-factor at line 10591 @> WARNING failed to parse beta-factor at line 10592 @> WARNING failed to parse beta-factor at line 10593 @> WARNING failed to parse beta-factor at line 10594 @> WARNING failed to parse beta-factor at line 10595 @> WARNING failed to parse beta-factor at line 10596 @> WARNING failed to parse beta-factor at line 10597 @> WARNING failed to parse beta-factor at line 10598 @> WARNING failed to parse beta-factor at line 10599 @> WARNING failed to parse beta-factor at line 10600 @> WARNING failed to parse beta-factor at line 10601 @> WARNING failed to parse beta-factor at line 10602 @> WARNING failed to parse beta-factor at line 10603 @> WARNING failed to parse beta-factor at line 10604 @> WARNING failed to parse beta-factor at line 10605 @> WARNING failed to parse beta-factor at line 10606 @> WARNING failed to parse beta-factor at line 10607 @> WARNING failed to parse beta-factor at line 10608 @> WARNING failed to parse beta-factor at line 10609 @> WARNING failed to parse beta-factor at line 10610 @> WARNING failed to parse beta-factor at line 10611 @> WARNING failed to parse beta-factor at line 10612 @> WARNING failed to parse beta-factor at line 10613 @> WARNING failed to parse beta-factor at line 10614 @> WARNING failed to parse beta-factor at line 10615 @> WARNING failed to parse beta-factor at line 10616 @> WARNING failed to parse beta-factor at line 10617 @> WARNING failed to parse beta-factor at line 10618 @> WARNING failed to parse beta-factor at line 10619 @> WARNING failed to parse beta-factor at line 10620 @> WARNING failed to parse beta-factor at line 10621 @> WARNING failed to parse beta-factor at line 10622 @> WARNING failed to parse beta-factor at line 10623 @> WARNING failed to parse beta-factor at line 10624 @> WARNING failed to parse beta-factor at line 10625 @> WARNING failed to parse beta-factor at line 10626 @> WARNING failed to parse beta-factor at line 10627 @> WARNING failed to parse beta-factor at line 10628 @> WARNING failed to parse beta-factor at line 10629 @> WARNING failed to parse beta-factor at line 10630 @> WARNING failed to parse beta-factor at line 10631 @> WARNING failed to parse beta-factor at line 10632 @> WARNING failed to parse beta-factor at line 10633 @> WARNING failed to parse beta-factor at line 10634 @> WARNING failed to parse beta-factor at line 10635 @> WARNING failed to parse beta-factor at line 10636 @> WARNING failed to parse beta-factor at line 10637 @> WARNING failed to parse beta-factor at line 10638 @> WARNING failed to parse beta-factor at line 10639 @> WARNING failed to parse beta-factor at line 10640 @> WARNING failed to parse beta-factor at line 10641 @> WARNING failed to parse beta-factor at line 10642 @> WARNING failed to parse beta-factor at line 10643 @> WARNING failed to parse beta-factor at line 10644 @> WARNING failed to parse beta-factor at line 10645 @> WARNING failed to parse beta-factor at line 10646 @> WARNING failed to parse beta-factor at line 10647 @> WARNING failed to parse beta-factor at line 10648 @> WARNING failed to parse beta-factor at line 10649 @> WARNING failed to parse beta-factor at line 10650 @> WARNING failed to parse beta-factor at line 10651 @> WARNING failed to parse beta-factor at line 10652 @> WARNING failed to parse beta-factor at line 10653 @> WARNING failed to parse beta-factor at line 10654 @> WARNING failed to parse beta-factor at line 10655 @> WARNING failed to parse beta-factor at line 10656 @> WARNING failed to parse beta-factor at line 10657 @> WARNING failed to parse beta-factor at line 10658 @> WARNING failed to parse beta-factor at line 10659 @> WARNING failed to parse beta-factor at line 10660 @> WARNING failed to parse beta-factor at line 10661 @> WARNING failed to parse beta-factor at line 10662 @> WARNING failed to parse beta-factor at line 10663 @> WARNING failed to parse beta-factor at line 10664 @> WARNING failed to parse beta-factor at line 10665 @> WARNING failed to parse beta-factor at line 10666 @> WARNING failed to parse beta-factor at line 10667 @> WARNING failed to parse beta-factor at line 10668 @> WARNING failed to parse beta-factor at line 10669 @> WARNING failed to parse beta-factor at line 10670 @> WARNING failed to parse beta-factor at line 10671 @> WARNING failed to parse beta-factor at line 10672 @> WARNING failed to parse beta-factor at line 10673 @> WARNING failed to parse beta-factor at line 10674 @> WARNING failed to parse beta-factor at line 10675 @> WARNING failed to parse beta-factor at line 10676 @> WARNING failed to parse beta-factor at line 10677 @> WARNING failed to parse beta-factor at line 10678 @> WARNING failed to parse beta-factor at line 10679 @> WARNING failed to parse beta-factor at line 10680 @> WARNING failed to parse beta-factor at line 10681 @> WARNING failed to parse beta-factor at line 10682 @> WARNING failed to parse beta-factor at line 10683 @> WARNING failed to parse beta-factor at line 10684 @> WARNING failed to parse beta-factor at line 10685 @> WARNING failed to parse beta-factor at line 10686 @> WARNING failed to parse beta-factor at line 10687 @> WARNING failed to parse beta-factor at line 10688 @> WARNING failed to parse beta-factor at line 10689 @> WARNING failed to parse beta-factor at line 10690 @> WARNING failed to parse beta-factor at line 10691 @> WARNING failed to parse beta-factor at line 10692 @> WARNING failed to parse beta-factor at line 10693 @> WARNING failed to parse beta-factor at line 10694 @> WARNING failed to parse beta-factor at line 10695 @> WARNING failed to parse beta-factor at line 10696 @> WARNING failed to parse beta-factor at line 10697 @> WARNING failed to parse beta-factor at line 10698 @> WARNING failed to parse beta-factor at line 10699 @> WARNING failed to parse beta-factor at line 10700 @> WARNING failed to parse beta-factor at line 10701 @> WARNING failed to parse beta-factor at line 10702 @> WARNING failed to parse beta-factor at line 10703 @> WARNING failed to parse beta-factor at line 10704 @> WARNING failed to parse beta-factor at line 10705 @> WARNING failed to parse beta-factor at line 10706 @> WARNING failed to parse beta-factor at line 10707 @> WARNING failed to parse beta-factor at line 10708 @> WARNING failed to parse beta-factor at line 10709 @> WARNING failed to parse beta-factor at line 10710 @> WARNING failed to parse beta-factor at line 10711 @> WARNING failed to parse beta-factor at line 10712 @> WARNING failed to parse beta-factor at line 10713 @> WARNING failed to parse beta-factor at line 10714 @> WARNING failed to parse beta-factor at line 10715 @> WARNING failed to parse beta-factor at line 10716 @> WARNING failed to parse beta-factor at line 10717 @> WARNING failed to parse beta-factor at line 10718 @> WARNING failed to parse beta-factor at line 10719 @> WARNING failed to parse beta-factor at line 10720 @> WARNING failed to parse beta-factor at line 10721 @> WARNING failed to parse beta-factor at line 10722 @> WARNING failed to parse beta-factor at line 10723 @> WARNING failed to parse beta-factor at line 10724 @> WARNING failed to parse beta-factor at line 10725 @> WARNING failed to parse beta-factor at line 10726 @> WARNING failed to parse beta-factor at line 10727 @> WARNING failed to parse beta-factor at line 10728 @> WARNING failed to parse beta-factor at line 10729 @> WARNING failed to parse beta-factor at line 10730 @> WARNING failed to parse beta-factor at line 10731 @> WARNING failed to parse beta-factor at line 10732 @> WARNING failed to parse beta-factor at line 10733 @> WARNING failed to parse beta-factor at line 10734 @> WARNING failed to parse beta-factor at line 10735 @> WARNING failed to parse beta-factor at line 10736 @> WARNING failed to parse beta-factor at line 10737 @> WARNING failed to parse beta-factor at line 10738 @> WARNING failed to parse beta-factor at line 10739 @> WARNING failed to parse beta-factor at line 10740 @> WARNING failed to parse beta-factor at line 10741 @> WARNING failed to parse beta-factor at line 10742 @> WARNING failed to parse beta-factor at line 10743 @> WARNING failed to parse beta-factor at line 10744 @> WARNING failed to parse beta-factor at line 10745 @> WARNING failed to parse beta-factor at line 10746 @> WARNING failed to parse beta-factor at line 10747 @> WARNING failed to parse beta-factor at line 10748 @> WARNING failed to parse beta-factor at line 10749 @> WARNING failed to parse beta-factor at line 10750 @> WARNING failed to parse beta-factor at line 10751 @> WARNING failed to parse beta-factor at line 10752 @> WARNING failed to parse beta-factor at line 10753 @> WARNING failed to parse beta-factor at line 10754 @> WARNING failed to parse beta-factor at line 10755 @> WARNING failed to parse beta-factor at line 10756 @> WARNING failed to parse beta-factor at line 10757 @> WARNING failed to parse beta-factor at line 10758 @> WARNING failed to parse beta-factor at line 10759 @> WARNING failed to parse beta-factor at line 10760 @> WARNING failed to parse beta-factor at line 10761 @> WARNING failed to parse beta-factor at line 10762 @> WARNING failed to parse beta-factor at line 10763 @> WARNING failed to parse beta-factor at line 10764 @> WARNING failed to parse beta-factor at line 10765 @> WARNING failed to parse beta-factor at line 10766 @> WARNING failed to parse beta-factor at line 10767 @> WARNING failed to parse beta-factor at line 10768 @> WARNING failed to parse beta-factor at line 10769 @> WARNING failed to parse beta-factor at line 10770 @> WARNING failed to parse beta-factor at line 10771 @> WARNING failed to parse beta-factor at line 10772 @> WARNING failed to parse beta-factor at line 10773 @> WARNING failed to parse beta-factor at line 10774 @> WARNING failed to parse beta-factor at line 10775 @> WARNING failed to parse beta-factor at line 10776 @> WARNING failed to parse beta-factor at line 10777 @> WARNING failed to parse beta-factor at line 10778 @> WARNING failed to parse beta-factor at line 10779 @> WARNING failed to parse beta-factor at line 10780 @> WARNING failed to parse beta-factor at line 10781 @> WARNING failed to parse beta-factor at line 10782 @> WARNING failed to parse beta-factor at line 10783 @> WARNING failed to parse beta-factor at line 10784 @> WARNING failed to parse beta-factor at line 10785 @> WARNING failed to parse beta-factor at line 10786 @> WARNING failed to parse beta-factor at line 10787 @> WARNING failed to parse beta-factor at line 10788 @> WARNING failed to parse beta-factor at line 10789 @> WARNING failed to parse beta-factor at line 10790 @> WARNING failed to parse beta-factor at line 10791 @> WARNING failed to parse beta-factor at line 10792 @> WARNING failed to parse beta-factor at line 10793 @> WARNING failed to parse beta-factor at line 10794 @> WARNING failed to parse beta-factor at line 10795 @> WARNING failed to parse beta-factor at line 10796 @> WARNING failed to parse beta-factor at line 10797 @> WARNING failed to parse beta-factor at line 10798 @> WARNING failed to parse beta-factor at line 10799 @> WARNING failed to parse beta-factor at line 10800 @> WARNING failed to parse beta-factor at line 10801 @> WARNING failed to parse beta-factor at line 10802 @> WARNING failed to parse beta-factor at line 10803 @> WARNING failed to parse beta-factor at line 10804 @> WARNING failed to parse beta-factor at line 10805 @> WARNING failed to parse beta-factor at line 10806 @> WARNING failed to parse beta-factor at line 10807 @> WARNING failed to parse beta-factor at line 10808 @> WARNING failed to parse beta-factor at line 10809 @> WARNING failed to parse beta-factor at line 10810 @> WARNING failed to parse beta-factor at line 10811 @> WARNING failed to parse beta-factor at line 10812 @> WARNING failed to parse beta-factor at line 10813 @> WARNING failed to parse beta-factor at line 10814 @> WARNING failed to parse beta-factor at line 10815 @> WARNING failed to parse beta-factor at line 10816 @> WARNING failed to parse beta-factor at line 10817 @> WARNING failed to parse beta-factor at line 10818 @> WARNING failed to parse beta-factor at line 10819 @> WARNING failed to parse beta-factor at line 10820 @> WARNING failed to parse beta-factor at line 10821 @> WARNING failed to parse beta-factor at line 10822 @> WARNING failed to parse beta-factor at line 10823 @> WARNING failed to parse beta-factor at line 10824 @> WARNING failed to parse beta-factor at line 10825 @> WARNING failed to parse beta-factor at line 10826 @> WARNING failed to parse beta-factor at line 10827 @> WARNING failed to parse beta-factor at line 10828 @> WARNING failed to parse beta-factor at line 10829 @> WARNING failed to parse beta-factor at line 10830 @> WARNING failed to parse beta-factor at line 10831 @> WARNING failed to parse beta-factor at line 10832 @> WARNING failed to parse beta-factor at line 10833 @> WARNING failed to parse beta-factor at line 10834 @> WARNING failed to parse beta-factor at line 10835 @> WARNING failed to parse beta-factor at line 10836 @> WARNING failed to parse beta-factor at line 10837 @> WARNING failed to parse beta-factor at line 10838 @> WARNING failed to parse beta-factor at line 10839 @> WARNING failed to parse beta-factor at line 10840 @> WARNING failed to parse beta-factor at line 10841 @> WARNING failed to parse beta-factor at line 10842 @> WARNING failed to parse beta-factor at line 10843 @> WARNING failed to parse beta-factor at line 10844 @> WARNING failed to parse beta-factor at line 10845 @> WARNING failed to parse beta-factor at line 10846 @> WARNING failed to parse beta-factor at line 10847 @> WARNING failed to parse beta-factor at line 10848 @> WARNING failed to parse beta-factor at line 10849 @> WARNING failed to parse beta-factor at line 10850 @> WARNING failed to parse beta-factor at line 10851 @> WARNING failed to parse beta-factor at line 10852 @> WARNING failed to parse beta-factor at line 10853 @> WARNING failed to parse beta-factor at line 10854 @> WARNING failed to parse beta-factor at line 10855 @> WARNING failed to parse beta-factor at line 10856 @> WARNING failed to parse beta-factor at line 10857 @> WARNING failed to parse beta-factor at line 10858 @> WARNING failed to parse beta-factor at line 10859 @> WARNING failed to parse beta-factor at line 10860 @> WARNING failed to parse beta-factor at line 10861 @> WARNING failed to parse beta-factor at line 10862 @> WARNING failed to parse beta-factor at line 10863 @> WARNING failed to parse beta-factor at line 10864 @> WARNING failed to parse beta-factor at line 10865 @> WARNING failed to parse beta-factor at line 10866 @> WARNING failed to parse beta-factor at line 10867 @> WARNING failed to parse beta-factor at line 10868 @> WARNING failed to parse beta-factor at line 10869 @> WARNING failed to parse beta-factor at line 10870 @> WARNING failed to parse beta-factor at line 10871 @> WARNING failed to parse beta-factor at line 10872 @> WARNING failed to parse beta-factor at line 10873 @> WARNING failed to parse beta-factor at line 10874 @> WARNING failed to parse beta-factor at line 10875 @> WARNING failed to parse beta-factor at line 10876 @> WARNING failed to parse beta-factor at line 10877 @> WARNING failed to parse beta-factor at line 10878 @> WARNING failed to parse beta-factor at line 10879 @> WARNING failed to parse beta-factor at line 10880 @> WARNING failed to parse beta-factor at line 10881 @> WARNING failed to parse beta-factor at line 10882 @> WARNING failed to parse beta-factor at line 10883 @> WARNING failed to parse beta-factor at line 10884 @> WARNING failed to parse beta-factor at line 10885 @> WARNING failed to parse beta-factor at line 10886 @> WARNING failed to parse beta-factor at line 10887 @> WARNING failed to parse beta-factor at line 10888 @> WARNING failed to parse beta-factor at line 10889 @> WARNING failed to parse beta-factor at line 10890 @> WARNING failed to parse beta-factor at line 10891 @> WARNING failed to parse beta-factor at line 10892 @> WARNING failed to parse beta-factor at line 10893 @> WARNING failed to parse beta-factor at line 10894 @> WARNING failed to parse beta-factor at line 10895 @> WARNING failed to parse beta-factor at line 10896 @> WARNING failed to parse beta-factor at line 10897 @> WARNING failed to parse beta-factor at line 10898 @> WARNING failed to parse beta-factor at line 10899 @> WARNING failed to parse beta-factor at line 10900 @> WARNING failed to parse beta-factor at line 10901 @> WARNING failed to parse beta-factor at line 10902 @> WARNING failed to parse beta-factor at line 10903 @> WARNING failed to parse beta-factor at line 10904 @> WARNING failed to parse beta-factor at line 10905 @> WARNING failed to parse beta-factor at line 10906 @> WARNING failed to parse beta-factor at line 10907 @> WARNING failed to parse beta-factor at line 10908 @> WARNING failed to parse beta-factor at line 10909 @> WARNING failed to parse beta-factor at line 10910 @> WARNING failed to parse beta-factor at line 10911 @> WARNING failed to parse beta-factor at line 10912 @> WARNING failed to parse beta-factor at line 10913 @> WARNING failed to parse beta-factor at line 10914 @> WARNING failed to parse beta-factor at line 10915 @> WARNING failed to parse beta-factor at line 10916 @> WARNING failed to parse beta-factor at line 10917 @> WARNING failed to parse beta-factor at line 10918 @> WARNING failed to parse beta-factor at line 10919 @> WARNING failed to parse beta-factor at line 10920 @> WARNING failed to parse beta-factor at line 10921 @> WARNING failed to parse beta-factor at line 10922 @> WARNING failed to parse beta-factor at line 10923 @> WARNING failed to parse beta-factor at line 10924 @> WARNING failed to parse beta-factor at line 10925 @> WARNING failed to parse beta-factor at line 10926 @> WARNING failed to parse beta-factor at line 10927 @> WARNING failed to parse beta-factor at line 10928 @> WARNING failed to parse beta-factor at line 10929 @> WARNING failed to parse beta-factor at line 10930 @> WARNING failed to parse beta-factor at line 10931 @> WARNING failed to parse beta-factor at line 10932 @> WARNING failed to parse beta-factor at line 10933 @> WARNING failed to parse beta-factor at line 10934 @> WARNING failed to parse beta-factor at line 10935 @> WARNING failed to parse beta-factor at line 10936 @> WARNING failed to parse beta-factor at line 10937 @> WARNING failed to parse beta-factor at line 10938 @> WARNING failed to parse beta-factor at line 10939 @> WARNING failed to parse beta-factor at line 10940 @> WARNING failed to parse beta-factor at line 10941 @> WARNING failed to parse beta-factor at line 10942 @> WARNING failed to parse beta-factor at line 10943 @> WARNING failed to parse beta-factor at line 10944 @> WARNING failed to parse beta-factor at line 10945 @> WARNING failed to parse beta-factor at line 10946 @> WARNING failed to parse beta-factor at line 10947 @> WARNING failed to parse beta-factor at line 10948 @> WARNING failed to parse beta-factor at line 10949 @> WARNING failed to parse beta-factor at line 10950 @> WARNING failed to parse beta-factor at line 10951 @> WARNING failed to parse beta-factor at line 10952 @> WARNING failed to parse beta-factor at line 10953 @> WARNING failed to parse beta-factor at line 10954 @> WARNING failed to parse beta-factor at line 10955 @> WARNING failed to parse beta-factor at line 10956 @> WARNING failed to parse beta-factor at line 10957 @> WARNING failed to parse beta-factor at line 10958 @> WARNING failed to parse beta-factor at line 10959 @> WARNING failed to parse beta-factor at line 10960 @> WARNING failed to parse beta-factor at line 10961 @> WARNING failed to parse beta-factor at line 10962 @> WARNING failed to parse beta-factor at line 10963 @> WARNING failed to parse beta-factor at line 10964 @> WARNING failed to parse beta-factor at line 10965 @> WARNING failed to parse beta-factor at line 10966 @> WARNING failed to parse beta-factor at line 10967 @> WARNING failed to parse beta-factor at line 10968 @> WARNING failed to parse beta-factor at line 10969 @> WARNING failed to parse beta-factor at line 10970 @> WARNING failed to parse beta-factor at line 10971 @> WARNING failed to parse beta-factor at line 10972 @> WARNING failed to parse beta-factor at line 10973 @> WARNING failed to parse beta-factor at line 10974 @> WARNING failed to parse beta-factor at line 10975 @> WARNING failed to parse beta-factor at line 10976 @> WARNING failed to parse beta-factor at line 10977 @> WARNING failed to parse beta-factor at line 10978 @> WARNING failed to parse beta-factor at line 10979 @> WARNING failed to parse beta-factor at line 10980 @> WARNING failed to parse beta-factor at line 10981 @> WARNING failed to parse beta-factor at line 10982 @> WARNING failed to parse beta-factor at line 10983 @> WARNING failed to parse beta-factor at line 10984 @> WARNING failed to parse beta-factor at line 10985 @> WARNING failed to parse beta-factor at line 10986 @> WARNING failed to parse beta-factor at line 10987 @> WARNING failed to parse beta-factor at line 10988 @> WARNING failed to parse beta-factor at line 10989 @> WARNING failed to parse beta-factor at line 10990 @> WARNING failed to parse beta-factor at line 10991 @> WARNING failed to parse beta-factor at line 10992 @> WARNING failed to parse beta-factor at line 10993 @> WARNING failed to parse beta-factor at line 10994 @> WARNING failed to parse beta-factor at line 10995 @> WARNING failed to parse beta-factor at line 10996 @> WARNING failed to parse beta-factor at line 10997 @> WARNING failed to parse beta-factor at line 10998 @> WARNING failed to parse beta-factor at line 10999 @> WARNING failed to parse beta-factor at line 11000 @> WARNING failed to parse beta-factor at line 11001 @> WARNING failed to parse beta-factor at line 11002 @> WARNING failed to parse beta-factor at line 11003 @> WARNING failed to parse beta-factor at line 11004 @> WARNING failed to parse beta-factor at line 11005 @> WARNING failed to parse beta-factor at line 11006 @> WARNING failed to parse beta-factor at line 11007 @> WARNING failed to parse beta-factor at line 11008 @> WARNING failed to parse beta-factor at line 11009 @> WARNING failed to parse beta-factor at line 11010 @> WARNING failed to parse beta-factor at line 11011 @> WARNING failed to parse beta-factor at line 11012 @> WARNING failed to parse beta-factor at line 11013 @> WARNING failed to parse beta-factor at line 11014 @> WARNING failed to parse beta-factor at line 11015 @> WARNING failed to parse beta-factor at line 11016 @> WARNING failed to parse beta-factor at line 11017 @> WARNING failed to parse beta-factor at line 11018 @> WARNING failed to parse beta-factor at line 11019 @> WARNING failed to parse beta-factor at line 11020 @> WARNING failed to parse beta-factor at line 11021 @> WARNING failed to parse beta-factor at line 11022 @> WARNING failed to parse beta-factor at line 11023 @> WARNING failed to parse beta-factor at line 11024 @> WARNING failed to parse beta-factor at line 11025 @> WARNING failed to parse beta-factor at line 11026 @> WARNING failed to parse beta-factor at line 11027 @> WARNING failed to parse beta-factor at line 11028 @> WARNING failed to parse beta-factor at line 11029 @> WARNING failed to parse beta-factor at line 11030 @> WARNING failed to parse beta-factor at line 11031 @> WARNING failed to parse beta-factor at line 11032 @> WARNING failed to parse beta-factor at line 11033 @> WARNING failed to parse beta-factor at line 11034 @> WARNING failed to parse beta-factor at line 11035 @> WARNING failed to parse beta-factor at line 11036 @> WARNING failed to parse beta-factor at line 11037 @> WARNING failed to parse beta-factor at line 11038 @> WARNING failed to parse beta-factor at line 11039 @> WARNING failed to parse beta-factor at line 11040 @> WARNING failed to parse beta-factor at line 11041 @> WARNING failed to parse beta-factor at line 11042 @> WARNING failed to parse beta-factor at line 11043 @> WARNING failed to parse beta-factor at line 11044 @> WARNING failed to parse beta-factor at line 11045 @> WARNING failed to parse beta-factor at line 11046 @> WARNING failed to parse beta-factor at line 11047 @> WARNING failed to parse beta-factor at line 11048 @> WARNING failed to parse beta-factor at line 11049 @> WARNING failed to parse beta-factor at line 11050 @> WARNING failed to parse beta-factor at line 11051 @> WARNING failed to parse beta-factor at line 11052 @> WARNING failed to parse beta-factor at line 11053 @> WARNING failed to parse beta-factor at line 11054 @> WARNING failed to parse beta-factor at line 11055 @> WARNING failed to parse beta-factor at line 11056 @> WARNING failed to parse beta-factor at line 11057 @> WARNING failed to parse beta-factor at line 11058 @> WARNING failed to parse beta-factor at line 11059 @> WARNING failed to parse beta-factor at line 11060 @> WARNING failed to parse beta-factor at line 11061 @> WARNING failed to parse beta-factor at line 11062 @> WARNING failed to parse beta-factor at line 11063 @> WARNING failed to parse beta-factor at line 11064 @> WARNING failed to parse beta-factor at line 11065 @> WARNING failed to parse beta-factor at line 11066 @> WARNING failed to parse beta-factor at line 11067 @> WARNING failed to parse beta-factor at line 11068 @> WARNING failed to parse beta-factor at line 11069 @> WARNING failed to parse beta-factor at line 11070 @> WARNING failed to parse beta-factor at line 11071 @> WARNING failed to parse beta-factor at line 11072 @> WARNING failed to parse beta-factor at line 11073 @> WARNING failed to parse beta-factor at line 11074 @> WARNING failed to parse beta-factor at line 11075 @> WARNING failed to parse beta-factor at line 11076 @> WARNING failed to parse beta-factor at line 11077 @> WARNING failed to parse beta-factor at line 11078 @> WARNING failed to parse beta-factor at line 11079 @> WARNING failed to parse beta-factor at line 11080 @> WARNING failed to parse beta-factor at line 11081 @> WARNING failed to parse beta-factor at line 11082 @> WARNING failed to parse beta-factor at line 11083 @> WARNING failed to parse beta-factor at line 11084 @> WARNING failed to parse beta-factor at line 11085 @> WARNING failed to parse beta-factor at line 11086 @> WARNING failed to parse beta-factor at line 11087 @> WARNING failed to parse beta-factor at line 11088 @> WARNING failed to parse beta-factor at line 11089 @> WARNING failed to parse beta-factor at line 11090 @> WARNING failed to parse beta-factor at line 11091 @> WARNING failed to parse beta-factor at line 11092 @> WARNING failed to parse beta-factor at line 11093 @> WARNING failed to parse beta-factor at line 11094 @> WARNING failed to parse beta-factor at line 11095 @> WARNING failed to parse beta-factor at line 11096 @> WARNING failed to parse beta-factor at line 11097 @> WARNING failed to parse beta-factor at line 11098 @> WARNING failed to parse beta-factor at line 11099 @> WARNING failed to parse beta-factor at line 11100 @> WARNING failed to parse beta-factor at line 11101 @> WARNING failed to parse beta-factor at line 11102 @> WARNING failed to parse beta-factor at line 11103 @> WARNING failed to parse beta-factor at line 11104 @> WARNING failed to parse beta-factor at line 11105 @> WARNING failed to parse beta-factor at line 11106 @> WARNING failed to parse beta-factor at line 11107 @> WARNING failed to parse beta-factor at line 11108 @> WARNING failed to parse beta-factor at line 11109 @> WARNING failed to parse beta-factor at line 11110 @> WARNING failed to parse beta-factor at line 11111 @> WARNING failed to parse beta-factor at line 11112 @> WARNING failed to parse beta-factor at line 11113 @> WARNING failed to parse beta-factor at line 11114 @> WARNING failed to parse beta-factor at line 11115 @> WARNING failed to parse beta-factor at line 11116 @> WARNING failed to parse beta-factor at line 11117 @> WARNING failed to parse beta-factor at line 11118 @> WARNING failed to parse beta-factor at line 11119 @> WARNING failed to parse beta-factor at line 11120 @> WARNING failed to parse beta-factor at line 11121 @> WARNING failed to parse beta-factor at line 11122 @> WARNING failed to parse beta-factor at line 11123 @> WARNING failed to parse beta-factor at line 11124 @> WARNING failed to parse beta-factor at line 11125 @> WARNING failed to parse beta-factor at line 11126 @> WARNING failed to parse beta-factor at line 11127 @> WARNING failed to parse beta-factor at line 11128 @> WARNING failed to parse beta-factor at line 11129 @> WARNING failed to parse beta-factor at line 11130 @> WARNING failed to parse beta-factor at line 11131 @> WARNING failed to parse beta-factor at line 11132 @> WARNING failed to parse beta-factor at line 11133 @> WARNING failed to parse beta-factor at line 11134 @> WARNING failed to parse beta-factor at line 11135 @> WARNING failed to parse beta-factor at line 11136 @> WARNING failed to parse beta-factor at line 11137 @> WARNING failed to parse beta-factor at line 11138 @> WARNING failed to parse beta-factor at line 11139 @> WARNING failed to parse beta-factor at line 11140 @> WARNING failed to parse beta-factor at line 11141 @> WARNING failed to parse beta-factor at line 11142 @> WARNING failed to parse beta-factor at line 11143 @> WARNING failed to parse beta-factor at line 11144 @> WARNING failed to parse beta-factor at line 11145 @> WARNING failed to parse beta-factor at line 11146 @> WARNING failed to parse beta-factor at line 11147 @> WARNING failed to parse beta-factor at line 11148 @> WARNING failed to parse beta-factor at line 11149 @> WARNING failed to parse beta-factor at line 11150 @> WARNING failed to parse beta-factor at line 11151 @> WARNING failed to parse beta-factor at line 11152 @> WARNING failed to parse beta-factor at line 11153 @> WARNING failed to parse beta-factor at line 11154 @> WARNING failed to parse beta-factor at line 11155 @> WARNING failed to parse beta-factor at line 11156 @> WARNING failed to parse beta-factor at line 11157 @> WARNING failed to parse beta-factor at line 11158 @> WARNING failed to parse beta-factor at line 11159 @> WARNING failed to parse beta-factor at line 11160 @> WARNING failed to parse beta-factor at line 11161 @> WARNING failed to parse beta-factor at line 11162 @> WARNING failed to parse beta-factor at line 11163 @> WARNING failed to parse beta-factor at line 11164 @> WARNING failed to parse beta-factor at line 11165 @> WARNING failed to parse beta-factor at line 11166 @> WARNING failed to parse beta-factor at line 11167 @> WARNING failed to parse beta-factor at line 11168 @> WARNING failed to parse beta-factor at line 11169 @> WARNING failed to parse beta-factor at line 11170 @> WARNING failed to parse beta-factor at line 11171 @> WARNING failed to parse beta-factor at line 11172 @> WARNING failed to parse beta-factor at line 11173 @> WARNING failed to parse beta-factor at line 11174 @> WARNING failed to parse beta-factor at line 11175 @> WARNING failed to parse beta-factor at line 11176 @> WARNING failed to parse beta-factor at line 11177 @> WARNING failed to parse beta-factor at line 11178 @> WARNING failed to parse beta-factor at line 11179 @> WARNING failed to parse beta-factor at line 11180 @> WARNING failed to parse beta-factor at line 11181 @> WARNING failed to parse beta-factor at line 11182 @> WARNING failed to parse beta-factor at line 11183 @> WARNING failed to parse beta-factor at line 11184 @> WARNING failed to parse beta-factor at line 11185 @> WARNING failed to parse beta-factor at line 11186 @> WARNING failed to parse beta-factor at line 11187 @> WARNING failed to parse beta-factor at line 11188 @> WARNING failed to parse beta-factor at line 11189 @> WARNING failed to parse beta-factor at line 11190 @> WARNING failed to parse beta-factor at line 11191 @> WARNING failed to parse beta-factor at line 11192 @> WARNING failed to parse beta-factor at line 11193 @> WARNING failed to parse beta-factor at line 11194 @> WARNING failed to parse beta-factor at line 11195 @> WARNING failed to parse beta-factor at line 11196 @> WARNING failed to parse beta-factor at line 11197 @> WARNING failed to parse beta-factor at line 11198 @> WARNING failed to parse beta-factor at line 11199 @> WARNING failed to parse beta-factor at line 11200 @> WARNING failed to parse beta-factor at line 11201 @> WARNING failed to parse beta-factor at line 11202 @> WARNING failed to parse beta-factor at line 11203 @> WARNING failed to parse beta-factor at line 11204 @> WARNING failed to parse beta-factor at line 11205 @> WARNING failed to parse beta-factor at line 11206 @> WARNING failed to parse beta-factor at line 11207 @> WARNING failed to parse beta-factor at line 11208 @> WARNING failed to parse beta-factor at line 11209 @> WARNING failed to parse beta-factor at line 11210 @> WARNING failed to parse beta-factor at line 11211 @> WARNING failed to parse beta-factor at line 11212 @> WARNING failed to parse beta-factor at line 11213 @> WARNING failed to parse beta-factor at line 11214 @> WARNING failed to parse beta-factor at line 11215 @> WARNING failed to parse beta-factor at line 11216 @> WARNING failed to parse beta-factor at line 11217 @> WARNING failed to parse beta-factor at line 11218 @> WARNING failed to parse beta-factor at line 11219 @> WARNING failed to parse beta-factor at line 11220 @> WARNING failed to parse beta-factor at line 11221 @> WARNING failed to parse beta-factor at line 11222 @> WARNING failed to parse beta-factor at line 11223 @> WARNING failed to parse beta-factor at line 11224 @> WARNING failed to parse beta-factor at line 11225 @> WARNING failed to parse beta-factor at line 11226 @> WARNING failed to parse beta-factor at line 11227 @> WARNING failed to parse beta-factor at line 11228 @> WARNING failed to parse beta-factor at line 11229 @> WARNING failed to parse beta-factor at line 11230 @> WARNING failed to parse beta-factor at line 11231 @> WARNING failed to parse beta-factor at line 11232 @> WARNING failed to parse beta-factor at line 11233 @> WARNING failed to parse beta-factor at line 11234 @> WARNING failed to parse beta-factor at line 11235 @> WARNING failed to parse beta-factor at line 11236 @> WARNING failed to parse beta-factor at line 11237 @> WARNING failed to parse beta-factor at line 11238 @> WARNING failed to parse beta-factor at line 11239 @> WARNING failed to parse beta-factor at line 11240 @> WARNING failed to parse beta-factor at line 11241 @> WARNING failed to parse beta-factor at line 11242 @> WARNING failed to parse beta-factor at line 11243 @> WARNING failed to parse beta-factor at line 11244 @> WARNING failed to parse beta-factor at line 11245 @> WARNING failed to parse beta-factor at line 11246 @> WARNING failed to parse beta-factor at line 11247 @> WARNING failed to parse beta-factor at line 11248 @> WARNING failed to parse beta-factor at line 11249 @> WARNING failed to parse beta-factor at line 11250 @> WARNING failed to parse beta-factor at line 11251 @> WARNING failed to parse beta-factor at line 11252 @> WARNING failed to parse beta-factor at line 11253 @> WARNING failed to parse beta-factor at line 11254 @> WARNING failed to parse beta-factor at line 11255 @> WARNING failed to parse beta-factor at line 11256 @> WARNING failed to parse beta-factor at line 11257 @> WARNING failed to parse beta-factor at line 11258 @> WARNING failed to parse beta-factor at line 11259 @> WARNING failed to parse beta-factor at line 11260 @> WARNING failed to parse beta-factor at line 11261 @> WARNING failed to parse beta-factor at line 11262 @> WARNING failed to parse beta-factor at line 11263 @> WARNING failed to parse beta-factor at line 11264 @> WARNING failed to parse beta-factor at line 11265 @> WARNING failed to parse beta-factor at line 11266 @> WARNING failed to parse beta-factor at line 11267 @> WARNING failed to parse beta-factor at line 11268 @> WARNING failed to parse beta-factor at line 11269 @> WARNING failed to parse beta-factor at line 11270 @> WARNING failed to parse beta-factor at line 11271 @> WARNING failed to parse beta-factor at line 11272 @> WARNING failed to parse beta-factor at line 11273 @> WARNING failed to parse beta-factor at line 11274 @> WARNING failed to parse beta-factor at line 11275 @> WARNING failed to parse beta-factor at line 11276 @> WARNING failed to parse beta-factor at line 11277 @> WARNING failed to parse beta-factor at line 11278 @> WARNING failed to parse beta-factor at line 11279 @> WARNING failed to parse beta-factor at line 11280 @> WARNING failed to parse beta-factor at line 11281 @> WARNING failed to parse beta-factor at line 11282 @> WARNING failed to parse beta-factor at line 11283 @> WARNING failed to parse beta-factor at line 11284 @> WARNING failed to parse beta-factor at line 11285 @> WARNING failed to parse beta-factor at line 11286 @> WARNING failed to parse beta-factor at line 11287 @> WARNING failed to parse beta-factor at line 11288 @> WARNING failed to parse beta-factor at line 11289 @> WARNING failed to parse beta-factor at line 11290 @> WARNING failed to parse beta-factor at line 11291 @> WARNING failed to parse beta-factor at line 11292 @> WARNING failed to parse beta-factor at line 11293 @> WARNING failed to parse beta-factor at line 11294 @> WARNING failed to parse beta-factor at line 11295 @> WARNING failed to parse beta-factor at line 11296 @> WARNING failed to parse beta-factor at line 11297 @> WARNING failed to parse beta-factor at line 11298 @> WARNING failed to parse beta-factor at line 11299 @> WARNING failed to parse beta-factor at line 11300 @> WARNING failed to parse beta-factor at line 11301 @> WARNING failed to parse beta-factor at line 11302 @> WARNING failed to parse beta-factor at line 11303 @> WARNING failed to parse beta-factor at line 11304 @> WARNING failed to parse beta-factor at line 11305 @> WARNING failed to parse beta-factor at line 11306 @> WARNING failed to parse beta-factor at line 11307 @> WARNING failed to parse beta-factor at line 11308 @> WARNING failed to parse beta-factor at line 11309 @> WARNING failed to parse beta-factor at line 11310 @> WARNING failed to parse beta-factor at line 11311 @> WARNING failed to parse beta-factor at line 11312 @> WARNING failed to parse beta-factor at line 11313 @> WARNING failed to parse beta-factor at line 11314 @> WARNING failed to parse beta-factor at line 11315 @> WARNING failed to parse beta-factor at line 11316 @> WARNING failed to parse beta-factor at line 11317 @> WARNING failed to parse beta-factor at line 11318 @> WARNING failed to parse beta-factor at line 11319 @> WARNING failed to parse beta-factor at line 11320 @> WARNING failed to parse beta-factor at line 11321 @> WARNING failed to parse beta-factor at line 11322 @> WARNING failed to parse beta-factor at line 11323 @> WARNING failed to parse beta-factor at line 11324 @> WARNING failed to parse beta-factor at line 11325 @> WARNING failed to parse beta-factor at line 11326 @> WARNING failed to parse beta-factor at line 11327 @> WARNING failed to parse beta-factor at line 11328 @> WARNING failed to parse beta-factor at line 11329 @> WARNING failed to parse beta-factor at line 11330 @> WARNING failed to parse beta-factor at line 11331 @> WARNING failed to parse beta-factor at line 11332 @> WARNING failed to parse beta-factor at line 11333 @> WARNING failed to parse beta-factor at line 11334 @> WARNING failed to parse beta-factor at line 11335 @> WARNING failed to parse beta-factor at line 11336 @> WARNING failed to parse beta-factor at line 11337 @> WARNING failed to parse beta-factor at line 11338 @> WARNING failed to parse beta-factor at line 11339 @> WARNING failed to parse beta-factor at line 11340 @> WARNING failed to parse beta-factor at line 11341 @> WARNING failed to parse beta-factor at line 11342 @> WARNING failed to parse beta-factor at line 11343 @> WARNING failed to parse beta-factor at line 11344 @> WARNING failed to parse beta-factor at line 11345 @> WARNING failed to parse beta-factor at line 11346 @> WARNING failed to parse beta-factor at line 11347 @> WARNING failed to parse beta-factor at line 11348 @> WARNING failed to parse beta-factor at line 11349 @> WARNING failed to parse beta-factor at line 11350 @> WARNING failed to parse beta-factor at line 11351 @> WARNING failed to parse beta-factor at line 11352 @> WARNING failed to parse beta-factor at line 11353 @> WARNING failed to parse beta-factor at line 11354 @> WARNING failed to parse beta-factor at line 11355 @> WARNING failed to parse beta-factor at line 11356 @> WARNING failed to parse beta-factor at line 11357 @> WARNING failed to parse beta-factor at line 11358 @> WARNING failed to parse beta-factor at line 11359 @> WARNING failed to parse beta-factor at line 11360 @> WARNING failed to parse beta-factor at line 11361 @> WARNING failed to parse beta-factor at line 11362 @> WARNING failed to parse beta-factor at line 11363 @> WARNING failed to parse beta-factor at line 11364 @> WARNING failed to parse beta-factor at line 11365 @> WARNING failed to parse beta-factor at line 11366 @> WARNING failed to parse beta-factor at line 11367 @> WARNING failed to parse beta-factor at line 11368 @> WARNING failed to parse beta-factor at line 11369 @> WARNING failed to parse beta-factor at line 11370 @> WARNING failed to parse beta-factor at line 11371 @> WARNING failed to parse beta-factor at line 11372 @> WARNING failed to parse beta-factor at line 11373 @> WARNING failed to parse beta-factor at line 11374 @> WARNING failed to parse beta-factor at line 11375 @> WARNING failed to parse beta-factor at line 11376 @> WARNING failed to parse beta-factor at line 11377 @> WARNING failed to parse beta-factor at line 11378 @> WARNING failed to parse beta-factor at line 11379 @> WARNING failed to parse beta-factor at line 11380 @> WARNING failed to parse beta-factor at line 11381 @> WARNING failed to parse beta-factor at line 11382 @> WARNING failed to parse beta-factor at line 11383 @> WARNING failed to parse beta-factor at line 11384 @> WARNING failed to parse beta-factor at line 11385 @> WARNING failed to parse beta-factor at line 11386 @> WARNING failed to parse beta-factor at line 11387 @> WARNING failed to parse beta-factor at line 11388 @> WARNING failed to parse beta-factor at line 11389 @> WARNING failed to parse beta-factor at line 11390 @> WARNING failed to parse beta-factor at line 11391 @> WARNING failed to parse beta-factor at line 11392 @> WARNING failed to parse beta-factor at line 11393 @> WARNING failed to parse beta-factor at line 11394 @> WARNING failed to parse beta-factor at line 11395 @> WARNING failed to parse beta-factor at line 11396 @> WARNING failed to parse beta-factor at line 11397 @> WARNING failed to parse beta-factor at line 11398 @> WARNING failed to parse beta-factor at line 11399 @> WARNING failed to parse beta-factor at line 11400 @> WARNING failed to parse beta-factor at line 11401 @> WARNING failed to parse beta-factor at line 11402 @> WARNING failed to parse beta-factor at line 11403 @> WARNING failed to parse beta-factor at line 11404 @> WARNING failed to parse beta-factor at line 11405 @> WARNING failed to parse beta-factor at line 11406 @> WARNING failed to parse beta-factor at line 11407 @> WARNING failed to parse beta-factor at line 11408 @> WARNING failed to parse beta-factor at line 11409 @> WARNING failed to parse beta-factor at line 11410 @> WARNING failed to parse beta-factor at line 11411 @> WARNING failed to parse beta-factor at line 11412 @> WARNING failed to parse beta-factor at line 11413 @> WARNING failed to parse beta-factor at line 11414 @> WARNING failed to parse beta-factor at line 11415 @> WARNING failed to parse beta-factor at line 11416 @> WARNING failed to parse beta-factor at line 11417 @> WARNING failed to parse beta-factor at line 11418 @> WARNING failed to parse beta-factor at line 11419 @> WARNING failed to parse beta-factor at line 11420 @> WARNING failed to parse beta-factor at line 11421 @> WARNING failed to parse beta-factor at line 11422 @> WARNING failed to parse beta-factor at line 11423 @> WARNING failed to parse beta-factor at line 11424 @> WARNING failed to parse beta-factor at line 11425 @> WARNING failed to parse beta-factor at line 11426 @> WARNING failed to parse beta-factor at line 11427 @> WARNING failed to parse beta-factor at line 11428 @> WARNING failed to parse beta-factor at line 11429 @> WARNING failed to parse beta-factor at line 11430 @> WARNING failed to parse beta-factor at line 11431 @> WARNING failed to parse beta-factor at line 11432 @> WARNING failed to parse beta-factor at line 11433 @> WARNING failed to parse beta-factor at line 11434 @> WARNING failed to parse beta-factor at line 11435 @> WARNING failed to parse beta-factor at line 11436 @> WARNING failed to parse beta-factor at line 11437 @> WARNING failed to parse beta-factor at line 11438 @> WARNING failed to parse beta-factor at line 11439 @> WARNING failed to parse beta-factor at line 11440 @> WARNING failed to parse beta-factor at line 11441 @> WARNING failed to parse beta-factor at line 11442 @> WARNING failed to parse beta-factor at line 11443 @> WARNING failed to parse beta-factor at line 11444 @> WARNING failed to parse beta-factor at line 11445 @> WARNING failed to parse beta-factor at line 11446 @> WARNING failed to parse beta-factor at line 11447 @> WARNING failed to parse beta-factor at line 11448 @> WARNING failed to parse beta-factor at line 11449 @> WARNING failed to parse beta-factor at line 11450 @> WARNING failed to parse beta-factor at line 11451 @> WARNING failed to parse beta-factor at line 11452 @> WARNING failed to parse beta-factor at line 11453 @> WARNING failed to parse beta-factor at line 11454 @> WARNING failed to parse beta-factor at line 11455 @> WARNING failed to parse beta-factor at line 11456 @> WARNING failed to parse beta-factor at line 11457 @> WARNING failed to parse beta-factor at line 11458 @> WARNING failed to parse beta-factor at line 11459 @> WARNING failed to parse beta-factor at line 11460 @> WARNING failed to parse beta-factor at line 11461 @> WARNING failed to parse beta-factor at line 11462 @> WARNING failed to parse beta-factor at line 11463 @> WARNING failed to parse beta-factor at line 11464 @> WARNING failed to parse beta-factor at line 11465 @> WARNING failed to parse beta-factor at line 11466 @> WARNING failed to parse beta-factor at line 11467 @> WARNING failed to parse beta-factor at line 11468 @> WARNING failed to parse beta-factor at line 11469 @> WARNING failed to parse beta-factor at line 11470 @> WARNING failed to parse beta-factor at line 11471 @> WARNING failed to parse beta-factor at line 11472 @> WARNING failed to parse beta-factor at line 11473 @> WARNING failed to parse beta-factor at line 11474 @> WARNING failed to parse beta-factor at line 11475 @> WARNING failed to parse beta-factor at line 11476 @> WARNING failed to parse beta-factor at line 11477 @> WARNING failed to parse beta-factor at line 11478 @> WARNING failed to parse beta-factor at line 11479 @> WARNING failed to parse beta-factor at line 11480 @> WARNING failed to parse beta-factor at line 11481 @> WARNING failed to parse beta-factor at line 11482 @> WARNING failed to parse beta-factor at line 11483 @> WARNING failed to parse beta-factor at line 11484 @> WARNING failed to parse beta-factor at line 11485 @> WARNING failed to parse beta-factor at line 11486 @> WARNING failed to parse beta-factor at line 11487 @> WARNING failed to parse beta-factor at line 11488 @> WARNING failed to parse beta-factor at line 11489 @> WARNING failed to parse beta-factor at line 11490 @> WARNING failed to parse beta-factor at line 11491 @> WARNING failed to parse beta-factor at line 11492 @> WARNING failed to parse beta-factor at line 11493 @> WARNING failed to parse beta-factor at line 11494 @> WARNING failed to parse beta-factor at line 11495 @> WARNING failed to parse beta-factor at line 11496 @> WARNING failed to parse beta-factor at line 11497 @> WARNING failed to parse beta-factor at line 11498 @> WARNING failed to parse beta-factor at line 11499 @> WARNING failed to parse beta-factor at line 11500 @> WARNING failed to parse beta-factor at line 11501 @> WARNING failed to parse beta-factor at line 11502 @> WARNING failed to parse beta-factor at line 11503 @> WARNING failed to parse beta-factor at line 11504 @> WARNING failed to parse beta-factor at line 11505 @> WARNING failed to parse beta-factor at line 11506 @> WARNING failed to parse beta-factor at line 11507 @> WARNING failed to parse beta-factor at line 11508 @> WARNING failed to parse beta-factor at line 11509 @> WARNING failed to parse beta-factor at line 11510 @> WARNING failed to parse beta-factor at line 11511 @> WARNING failed to parse beta-factor at line 11512 @> WARNING failed to parse beta-factor at line 11513 @> WARNING failed to parse beta-factor at line 11514 @> WARNING failed to parse beta-factor at line 11515 @> WARNING failed to parse beta-factor at line 11516 @> WARNING failed to parse beta-factor at line 11517 @> WARNING failed to parse beta-factor at line 11518 @> WARNING failed to parse beta-factor at line 11519 @> WARNING failed to parse beta-factor at line 11520 @> WARNING failed to parse beta-factor at line 11521 @> WARNING failed to parse beta-factor at line 11522 @> WARNING failed to parse beta-factor at line 11523 @> WARNING failed to parse beta-factor at line 11524 @> WARNING failed to parse beta-factor at line 11525 @> WARNING failed to parse beta-factor at line 11526 @> WARNING failed to parse beta-factor at line 11527 @> WARNING failed to parse beta-factor at line 11528 @> WARNING failed to parse beta-factor at line 11529 @> WARNING failed to parse beta-factor at line 11530 @> WARNING failed to parse beta-factor at line 11531 @> WARNING failed to parse beta-factor at line 11532 @> WARNING failed to parse beta-factor at line 11533 @> WARNING failed to parse beta-factor at line 11534 @> WARNING failed to parse beta-factor at line 11535 @> WARNING failed to parse beta-factor at line 11536 @> WARNING failed to parse beta-factor at line 11537 @> WARNING failed to parse beta-factor at line 11538 @> WARNING failed to parse beta-factor at line 11539 @> WARNING failed to parse beta-factor at line 11540 @> WARNING failed to parse beta-factor at line 11541 @> WARNING failed to parse beta-factor at line 11542 @> WARNING failed to parse beta-factor at line 11543 @> WARNING failed to parse beta-factor at line 11544 @> WARNING failed to parse beta-factor at line 11545 @> WARNING failed to parse beta-factor at line 11546 @> WARNING failed to parse beta-factor at line 11547 @> WARNING failed to parse beta-factor at line 11548 @> WARNING failed to parse beta-factor at line 11549 @> WARNING failed to parse beta-factor at line 11550 @> WARNING failed to parse beta-factor at line 11551 @> WARNING failed to parse beta-factor at line 11552 @> WARNING failed to parse beta-factor at line 11553 @> WARNING failed to parse beta-factor at line 11554 @> WARNING failed to parse beta-factor at line 11555 @> WARNING failed to parse beta-factor at line 11556 @> WARNING failed to parse beta-factor at line 11557 @> WARNING failed to parse beta-factor at line 11558 @> WARNING failed to parse beta-factor at line 11559 @> WARNING failed to parse beta-factor at line 11560 @> WARNING failed to parse beta-factor at line 11561 @> WARNING failed to parse beta-factor at line 11562 @> WARNING failed to parse beta-factor at line 11563 @> WARNING failed to parse beta-factor at line 11564 @> WARNING failed to parse beta-factor at line 11565 @> WARNING failed to parse beta-factor at line 11566 @> WARNING failed to parse beta-factor at line 11567 @> WARNING failed to parse beta-factor at line 11568 @> WARNING failed to parse beta-factor at line 11569 @> WARNING failed to parse beta-factor at line 11570 @> WARNING failed to parse beta-factor at line 11571 @> WARNING failed to parse beta-factor at line 11572 @> WARNING failed to parse beta-factor at line 11573 @> WARNING failed to parse beta-factor at line 11574 @> WARNING failed to parse beta-factor at line 11575 @> WARNING failed to parse beta-factor at line 11576 @> WARNING failed to parse beta-factor at line 11577 @> WARNING failed to parse beta-factor at line 11578 @> WARNING failed to parse beta-factor at line 11579 @> WARNING failed to parse beta-factor at line 11580 @> WARNING failed to parse beta-factor at line 11581 @> WARNING failed to parse beta-factor at line 11582 @> WARNING failed to parse beta-factor at line 11583 @> WARNING failed to parse beta-factor at line 11584 @> WARNING failed to parse beta-factor at line 11585 @> WARNING failed to parse beta-factor at line 11586 @> WARNING failed to parse beta-factor at line 11587 @> WARNING failed to parse beta-factor at line 11588 @> WARNING failed to parse beta-factor at line 11589 @> WARNING failed to parse beta-factor at line 11590 @> WARNING failed to parse beta-factor at line 11591 @> WARNING failed to parse beta-factor at line 11592 @> WARNING failed to parse beta-factor at line 11593 @> WARNING failed to parse beta-factor at line 11594 @> WARNING failed to parse beta-factor at line 11595 @> WARNING failed to parse beta-factor at line 11596 @> WARNING failed to parse beta-factor at line 11597 @> WARNING failed to parse beta-factor at line 11598 @> WARNING failed to parse beta-factor at line 11599 @> WARNING failed to parse beta-factor at line 11600 @> WARNING failed to parse beta-factor at line 11601 @> WARNING failed to parse beta-factor at line 11602 @> WARNING failed to parse beta-factor at line 11603 @> WARNING failed to parse beta-factor at line 11604 @> WARNING failed to parse beta-factor at line 11605 @> WARNING failed to parse beta-factor at line 11606 @> WARNING failed to parse beta-factor at line 11607 @> WARNING failed to parse beta-factor at line 11608 @> WARNING failed to parse beta-factor at line 11609 @> WARNING failed to parse beta-factor at line 11610 @> WARNING failed to parse beta-factor at line 11611 @> WARNING failed to parse beta-factor at line 11612 @> WARNING failed to parse beta-factor at line 11613 @> WARNING failed to parse beta-factor at line 11614 @> WARNING failed to parse beta-factor at line 11615 @> WARNING failed to parse beta-factor at line 11616 @> WARNING failed to parse beta-factor at line 11617 @> WARNING failed to parse beta-factor at line 11618 @> WARNING failed to parse beta-factor at line 11619 @> WARNING failed to parse beta-factor at line 11620 @> WARNING failed to parse beta-factor at line 11621 @> WARNING failed to parse beta-factor at line 11622 @> WARNING failed to parse beta-factor at line 11623 @> WARNING failed to parse beta-factor at line 11624 @> WARNING failed to parse beta-factor at line 11625 @> WARNING failed to parse beta-factor at line 11626 @> WARNING failed to parse beta-factor at line 11627 @> WARNING failed to parse beta-factor at line 11628 @> WARNING failed to parse beta-factor at line 11629 @> WARNING failed to parse beta-factor at line 11630 @> WARNING failed to parse beta-factor at line 11631 @> WARNING failed to parse beta-factor at line 11632 @> WARNING failed to parse beta-factor at line 11633 @> WARNING failed to parse beta-factor at line 11634 @> WARNING failed to parse beta-factor at line 11635 @> WARNING failed to parse beta-factor at line 11636 @> WARNING failed to parse beta-factor at line 11637 @> WARNING failed to parse beta-factor at line 11638 @> WARNING failed to parse beta-factor at line 11639 @> WARNING failed to parse beta-factor at line 11640 @> WARNING failed to parse beta-factor at line 11641 @> WARNING failed to parse beta-factor at line 11642 @> WARNING failed to parse beta-factor at line 11643 @> WARNING failed to parse beta-factor at line 11644 @> WARNING failed to parse beta-factor at line 11645 @> WARNING failed to parse beta-factor at line 11646 @> WARNING failed to parse beta-factor at line 11647 @> WARNING failed to parse beta-factor at line 11648 @> WARNING failed to parse beta-factor at line 11649 @> WARNING failed to parse beta-factor at line 11650 @> WARNING failed to parse beta-factor at line 11651 @> WARNING failed to parse beta-factor at line 11652 @> WARNING failed to parse beta-factor at line 11653 @> WARNING failed to parse beta-factor at line 11654 @> WARNING failed to parse beta-factor at line 11655 @> WARNING failed to parse beta-factor at line 11656 @> WARNING failed to parse beta-factor at line 11657 @> WARNING failed to parse beta-factor at line 11658 @> WARNING failed to parse beta-factor at line 11659 @> WARNING failed to parse beta-factor at line 11660 @> WARNING failed to parse beta-factor at line 11661 @> WARNING failed to parse beta-factor at line 11662 @> WARNING failed to parse beta-factor at line 11663 @> WARNING failed to parse beta-factor at line 11664 @> WARNING failed to parse beta-factor at line 11665 @> WARNING failed to parse beta-factor at line 11666 @> WARNING failed to parse beta-factor at line 11667 @> WARNING failed to parse beta-factor at line 11668 @> WARNING failed to parse beta-factor at line 11669 @> WARNING failed to parse beta-factor at line 11670 @> WARNING failed to parse beta-factor at line 11671 @> WARNING failed to parse beta-factor at line 11672 @> WARNING failed to parse beta-factor at line 11673 @> WARNING failed to parse beta-factor at line 11674 @> WARNING failed to parse beta-factor at line 11675 @> WARNING failed to parse beta-factor at line 11676 @> WARNING failed to parse beta-factor at line 11677 @> WARNING failed to parse beta-factor at line 11678 @> WARNING failed to parse beta-factor at line 11679 @> WARNING failed to parse beta-factor at line 11680 @> WARNING failed to parse beta-factor at line 11681 @> WARNING failed to parse beta-factor at line 11682 @> WARNING failed to parse beta-factor at line 11683 @> WARNING failed to parse beta-factor at line 11684 @> WARNING failed to parse beta-factor at line 11685 @> WARNING failed to parse beta-factor at line 11686 @> WARNING failed to parse beta-factor at line 11687 @> WARNING failed to parse beta-factor at line 11688 @> WARNING failed to parse beta-factor at line 11689 @> WARNING failed to parse beta-factor at line 11690 @> WARNING failed to parse beta-factor at line 11691 @> WARNING failed to parse beta-factor at line 11692 @> WARNING failed to parse beta-factor at line 11693 @> WARNING failed to parse beta-factor at line 11694 @> WARNING failed to parse beta-factor at line 11695 @> WARNING failed to parse beta-factor at line 11696 @> WARNING failed to parse beta-factor at line 11697 @> WARNING failed to parse beta-factor at line 11698 @> WARNING failed to parse beta-factor at line 11699 @> WARNING failed to parse beta-factor at line 11700 @> WARNING failed to parse beta-factor at line 11701 @> WARNING failed to parse beta-factor at line 11702 @> WARNING failed to parse beta-factor at line 11703 @> WARNING failed to parse beta-factor at line 11704 @> WARNING failed to parse beta-factor at line 11705 @> WARNING failed to parse beta-factor at line 11706 @> WARNING failed to parse beta-factor at line 11707 @> WARNING failed to parse beta-factor at line 11708 @> WARNING failed to parse beta-factor at line 11709 @> WARNING failed to parse beta-factor at line 11710 @> WARNING failed to parse beta-factor at line 11711 @> WARNING failed to parse beta-factor at line 11712 @> WARNING failed to parse beta-factor at line 11713 @> WARNING failed to parse beta-factor at line 11714 @> WARNING failed to parse beta-factor at line 11715 @> WARNING failed to parse beta-factor at line 11716 @> WARNING failed to parse beta-factor at line 11717 @> WARNING failed to parse beta-factor at line 11718 @> WARNING failed to parse beta-factor at line 11719 @> WARNING failed to parse beta-factor at line 11720 @> WARNING failed to parse beta-factor at line 11721 @> WARNING failed to parse beta-factor at line 11722 @> WARNING failed to parse beta-factor at line 11723 @> WARNING failed to parse beta-factor at line 11724 @> WARNING failed to parse beta-factor at line 11725 @> WARNING failed to parse beta-factor at line 11726 @> WARNING failed to parse beta-factor at line 11727 @> WARNING failed to parse beta-factor at line 11728 @> WARNING failed to parse beta-factor at line 11729 IOPub message rate exceeded. The notebook server will temporarily stop sending output to the client in order to avoid crashing it. To change this limit, set the config variable `--NotebookApp.iopub_msg_rate_limit`. Current values: NotebookApp.iopub_msg_rate_limit=1000.0 (msgs/sec) NotebookApp.rate_limit_window=3.0 (secs) @> WARNING failed to parse beta-factor at line 9628 @> WARNING failed to parse beta-factor at line 9629 @> WARNING failed to parse beta-factor at line 9630 @> WARNING failed to parse beta-factor at line 9631 @> WARNING failed to parse beta-factor at line 9632 @> WARNING failed to parse beta-factor at line 9633 @> WARNING failed to parse beta-factor at line 9634 @> WARNING failed to parse beta-factor at line 9635 @> WARNING failed to parse beta-factor at line 9636 @> WARNING failed to parse beta-factor at line 9637 @> WARNING failed to parse beta-factor at line 9638 @> WARNING failed to parse beta-factor at line 9639 @> WARNING failed to parse beta-factor at line 9640 @> WARNING failed to parse beta-factor at line 9641 @> WARNING failed to parse beta-factor at line 9642 @> WARNING failed to parse beta-factor at line 9643 @> WARNING failed to parse beta-factor at line 9644 @> WARNING failed to parse beta-factor at line 9645 @> WARNING failed to parse beta-factor at line 9646 @> WARNING failed to parse beta-factor at line 9647 @> WARNING failed to parse beta-factor at line 9648 @> WARNING failed to parse beta-factor at line 9649 @> WARNING failed to parse beta-factor at line 9650 @> WARNING failed to parse beta-factor at line 9651 @> WARNING failed to parse beta-factor at line 9652 @> WARNING failed to parse beta-factor at line 9653 @> WARNING failed to parse beta-factor at line 9654 @> WARNING failed to parse beta-factor at line 9655 @> WARNING failed to parse beta-factor at line 9656 @> WARNING failed to parse beta-factor at line 9657 @> WARNING failed to parse beta-factor at line 9658 @> WARNING failed to parse beta-factor at line 9659 @> WARNING failed to parse beta-factor at line 9660 @> WARNING failed to parse beta-factor at line 9661 @> WARNING failed to parse beta-factor at line 9662 @> WARNING failed to parse beta-factor at line 9663 @> WARNING failed to parse beta-factor at line 9664 @> WARNING failed to parse beta-factor at line 9665 @> WARNING failed to parse beta-factor at line 9666 @> WARNING failed to parse beta-factor at line 9667 @> WARNING failed to parse beta-factor at line 9668 @> WARNING failed to parse beta-factor at line 9669 @> WARNING failed to parse beta-factor at line 9670 @> WARNING failed to parse beta-factor at line 9671 @> WARNING failed to parse beta-factor at line 9672 @> WARNING failed to parse beta-factor at line 9673 @> WARNING failed to parse beta-factor at line 9674 @> WARNING failed to parse beta-factor at line 9675 @> WARNING failed to parse beta-factor at line 9676 @> WARNING failed to parse beta-factor at line 9677 @> WARNING failed to parse beta-factor at line 9678 @> WARNING failed to parse beta-factor at line 9679 @> WARNING failed to parse beta-factor at line 9680 @> WARNING failed to parse beta-factor at line 9681 @> WARNING failed to parse beta-factor at line 9682 @> WARNING failed to parse beta-factor at line 9683 @> WARNING failed to parse beta-factor at line 9684 @> WARNING failed to parse beta-factor at line 9685 @> WARNING failed to parse beta-factor at line 9686 @> WARNING failed to parse beta-factor at line 9687 @> WARNING failed to parse beta-factor at line 9688 @> WARNING failed to parse beta-factor at line 9689 @> WARNING failed to parse beta-factor at line 9690 @> WARNING failed to parse beta-factor at line 9691 @> WARNING failed to parse beta-factor at line 9692 @> WARNING failed to parse beta-factor at line 9693 @> WARNING failed to parse beta-factor at line 9694 @> WARNING failed to parse beta-factor at line 9695 @> WARNING failed to parse beta-factor at line 9696 @> WARNING failed to parse beta-factor at line 9697 @> WARNING failed to parse beta-factor at line 9698 @> WARNING failed to parse beta-factor at line 9699 @> WARNING failed to parse beta-factor at line 9700 @> WARNING failed to parse beta-factor at line 9701 @> WARNING failed to parse beta-factor at line 9702 @> WARNING failed to parse beta-factor at line 9703 @> WARNING failed to parse beta-factor at line 9704 @> WARNING failed to parse beta-factor at line 9705 @> WARNING failed to parse beta-factor at line 9706 @> WARNING failed to parse beta-factor at line 9707 @> WARNING failed to parse beta-factor at line 9708 @> WARNING failed to parse beta-factor at line 9709 @> WARNING failed to parse beta-factor at line 9710 @> WARNING failed to parse beta-factor at line 9711 @> WARNING failed to parse beta-factor at line 9712 @> WARNING failed to parse beta-factor at line 9713 @> WARNING failed to parse beta-factor at line 9714 @> WARNING failed to parse beta-factor at line 9715 @> WARNING failed to parse beta-factor at line 9716 @> WARNING failed to parse beta-factor at line 9717 @> WARNING failed to parse beta-factor at line 9718 @> WARNING failed to parse beta-factor at line 9719 @> WARNING failed to parse beta-factor at line 9720 @> WARNING failed to parse beta-factor at line 9721 @> WARNING failed to parse beta-factor at line 9722 @> WARNING failed to parse beta-factor at line 9723 @> WARNING failed to parse beta-factor at line 9724 @> WARNING failed to parse beta-factor at line 9725 @> WARNING failed to parse beta-factor at line 9726 @> WARNING failed to parse beta-factor at line 9727 @> WARNING failed to parse beta-factor at line 9728 @> WARNING failed to parse beta-factor at line 9729 @> WARNING failed to parse beta-factor at line 9730 @> WARNING failed to parse beta-factor at line 9731 @> WARNING failed to parse beta-factor at line 9732 @> WARNING failed to parse beta-factor at line 9733 @> WARNING failed to parse beta-factor at line 9734 @> WARNING failed to parse beta-factor at line 9735 @> WARNING failed to parse beta-factor at line 9736 @> WARNING failed to parse beta-factor at line 9737 @> WARNING failed to parse beta-factor at line 9738 @> WARNING failed to parse beta-factor at line 9739 @> WARNING failed to parse beta-factor at line 9740 @> WARNING failed to parse beta-factor at line 9741 @> WARNING failed to parse beta-factor at line 9742 @> WARNING failed to parse beta-factor at line 9743 @> WARNING failed to parse beta-factor at line 9744 @> WARNING failed to parse beta-factor at line 9745 @> WARNING failed to parse beta-factor at line 9746 @> WARNING failed to parse beta-factor at line 9747 @> WARNING failed to parse beta-factor at line 9748 @> WARNING failed to parse beta-factor at line 9749 @> WARNING failed to parse beta-factor at line 9750 @> WARNING failed to parse beta-factor at line 9751 @> WARNING failed to parse beta-factor at line 9752 @> WARNING failed to parse beta-factor at line 9753 @> WARNING failed to parse beta-factor at line 9754 @> WARNING failed to parse beta-factor at line 9755 @> WARNING failed to parse beta-factor at line 9756 @> WARNING failed to parse beta-factor at line 9757 @> WARNING failed to parse beta-factor at line 9758 @> WARNING failed to parse beta-factor at line 9759 @> WARNING failed to parse beta-factor at line 9760 @> WARNING failed to parse beta-factor at line 9761 @> WARNING failed to parse beta-factor at line 9762 @> WARNING failed to parse beta-factor at line 9763 @> WARNING failed to parse beta-factor at line 9764 @> WARNING failed to parse beta-factor at line 9765 @> WARNING failed to parse beta-factor at line 9766 @> WARNING failed to parse beta-factor at line 9767 @> WARNING failed to parse beta-factor at line 9768 @> WARNING failed to parse beta-factor at line 9769 @> WARNING failed to parse beta-factor at line 9770 @> WARNING failed to parse beta-factor at line 9771 @> WARNING failed to parse beta-factor at line 9772 @> WARNING failed to parse beta-factor at line 9773 @> WARNING failed to parse beta-factor at line 9774 @> WARNING failed to parse beta-factor at line 9775 @> WARNING failed to parse beta-factor at line 9776 @> WARNING failed to parse beta-factor at line 9777 @> WARNING failed to parse beta-factor at line 9778 @> WARNING failed to parse beta-factor at line 9779 @> WARNING failed to parse beta-factor at line 9780 @> WARNING failed to parse beta-factor at line 9781 @> WARNING failed to parse beta-factor at line 9782 @> WARNING failed to parse beta-factor at line 9783 @> WARNING failed to parse beta-factor at line 9784 @> WARNING failed to parse beta-factor at line 9785 @> WARNING failed to parse beta-factor at line 9786 @> WARNING failed to parse beta-factor at line 9787 @> WARNING failed to parse beta-factor at line 9788 @> WARNING failed to parse beta-factor at line 9789 @> WARNING failed to parse beta-factor at line 9790 @> WARNING failed to parse beta-factor at line 9791 @> WARNING failed to parse beta-factor at line 9792 @> WARNING failed to parse beta-factor at line 9793 @> WARNING failed to parse beta-factor at line 9794 @> WARNING failed to parse beta-factor at line 9795 @> WARNING failed to parse beta-factor at line 9796 @> WARNING failed to parse beta-factor at line 9797 @> WARNING failed to parse beta-factor at line 9798 @> WARNING failed to parse beta-factor at line 9799 @> WARNING failed to parse beta-factor at line 9800 @> WARNING failed to parse beta-factor at line 9801 @> WARNING failed to parse beta-factor at line 9802 @> WARNING failed to parse beta-factor at line 9803 @> WARNING failed to parse beta-factor at line 9804 @> WARNING failed to parse beta-factor at line 9805 @> WARNING failed to parse beta-factor at line 9806 @> WARNING failed to parse beta-factor at line 9807 @> WARNING failed to parse beta-factor at line 9808 @> WARNING failed to parse beta-factor at line 9809 @> WARNING failed to parse beta-factor at line 9810 @> WARNING failed to parse beta-factor at line 9811 @> WARNING failed to parse beta-factor at line 9812 @> WARNING failed to parse beta-factor at line 9813 @> WARNING failed to parse beta-factor at line 9814 @> WARNING failed to parse beta-factor at line 9815 @> WARNING failed to parse beta-factor at line 9816 @> WARNING failed to parse beta-factor at line 9817 @> WARNING failed to parse beta-factor at line 9818 @> WARNING failed to parse beta-factor at line 9819 @> WARNING failed to parse beta-factor at line 9820 @> WARNING failed to parse beta-factor at line 9821 @> WARNING failed to parse beta-factor at line 9822 @> WARNING failed to parse beta-factor at line 9823 @> WARNING failed to parse beta-factor at line 9824 @> WARNING failed to parse beta-factor at line 9825 @> WARNING failed to parse beta-factor at line 9826 @> WARNING failed to parse beta-factor at line 9827 @> WARNING failed to parse beta-factor at line 9828 @> WARNING failed to parse beta-factor at line 9829 @> WARNING failed to parse beta-factor at line 9830 @> WARNING failed to parse beta-factor at line 9831 @> WARNING failed to parse beta-factor at line 9832 @> WARNING failed to parse beta-factor at line 9833 @> WARNING failed to parse beta-factor at line 9834 @> WARNING failed to parse beta-factor at line 9835 @> WARNING failed to parse beta-factor at line 9836 @> WARNING failed to parse beta-factor at line 9837 @> WARNING failed to parse beta-factor at line 9838 @> WARNING failed to parse beta-factor at line 9839 @> WARNING failed to parse beta-factor at line 9840 @> WARNING failed to parse beta-factor at line 9841 @> WARNING failed to parse beta-factor at line 9842 @> WARNING failed to parse beta-factor at line 9843 @> WARNING failed to parse beta-factor at line 9844 @> WARNING failed to parse beta-factor at line 9845 @> WARNING failed to parse beta-factor at line 9846 @> WARNING failed to parse beta-factor at line 9847 @> WARNING failed to parse beta-factor at line 9848 @> WARNING failed to parse beta-factor at line 9849 @> WARNING failed to parse beta-factor at line 9850 @> WARNING failed to parse beta-factor at line 9851 @> WARNING failed to parse beta-factor at line 9852 @> WARNING failed to parse beta-factor at line 9853 @> WARNING failed to parse beta-factor at line 9854 @> WARNING failed to parse beta-factor at line 9855 @> WARNING failed to parse beta-factor at line 9856 @> WARNING failed to parse beta-factor at line 9857 @> WARNING failed to parse beta-factor at line 9858 @> WARNING failed to parse beta-factor at line 9859 @> WARNING failed to parse beta-factor at line 9860 @> WARNING failed to parse beta-factor at line 9861 @> WARNING failed to parse beta-factor at line 9862 @> WARNING failed to parse beta-factor at line 9863 @> WARNING failed to parse beta-factor at line 9864 @> WARNING failed to parse beta-factor at line 9865 @> WARNING failed to parse beta-factor at line 9866 @> WARNING failed to parse beta-factor at line 9867 @> WARNING failed to parse beta-factor at line 9868 @> WARNING failed to parse beta-factor at line 9869 @> WARNING failed to parse beta-factor at line 9870 @> WARNING failed to parse beta-factor at line 9871 @> WARNING failed to parse beta-factor at line 9872 @> WARNING failed to parse beta-factor at line 9873 @> WARNING failed to parse beta-factor at line 9874 @> WARNING failed to parse beta-factor at line 9875 @> WARNING failed to parse beta-factor at line 9876 @> WARNING failed to parse beta-factor at line 9877 @> WARNING failed to parse beta-factor at line 9878 @> WARNING failed to parse beta-factor at line 9879 @> WARNING failed to parse beta-factor at line 9880 @> WARNING failed to parse beta-factor at line 9881 @> WARNING failed to parse beta-factor at line 9882 @> WARNING failed to parse beta-factor at line 9883 @> WARNING failed to parse beta-factor at line 9884 @> WARNING failed to parse beta-factor at line 9885 @> WARNING failed to parse beta-factor at line 9886 @> WARNING failed to parse beta-factor at line 9887 @> WARNING failed to parse beta-factor at line 9888 @> WARNING failed to parse beta-factor at line 9889 @> WARNING failed to parse beta-factor at line 9890 @> WARNING failed to parse beta-factor at line 9891 @> WARNING failed to parse beta-factor at line 9892 @> WARNING failed to parse beta-factor at line 9893 @> WARNING failed to parse beta-factor at line 9894 @> WARNING failed to parse beta-factor at line 9895 @> WARNING failed to parse beta-factor at line 9896 @> WARNING failed to parse beta-factor at line 9897 @> WARNING failed to parse beta-factor at line 9898 @> WARNING failed to parse beta-factor at line 9899 @> WARNING failed to parse beta-factor at line 9900 @> WARNING failed to parse beta-factor at line 9901 @> WARNING failed to parse beta-factor at line 9902 @> WARNING failed to parse beta-factor at line 9903 @> WARNING failed to parse beta-factor at line 9904 @> WARNING failed to parse beta-factor at line 9905 @> WARNING failed to parse beta-factor at line 9906 @> WARNING failed to parse beta-factor at line 9907 @> WARNING failed to parse beta-factor at line 9908 @> WARNING failed to parse beta-factor at line 9909 @> WARNING failed to parse beta-factor at line 9910 @> WARNING failed to parse beta-factor at line 9911 @> WARNING failed to parse beta-factor at line 9912 @> WARNING failed to parse beta-factor at line 9913 @> WARNING failed to parse beta-factor at line 9914 @> WARNING failed to parse beta-factor at line 9915 @> WARNING failed to parse beta-factor at line 9916 @> WARNING failed to parse beta-factor at line 9917 @> WARNING failed to parse beta-factor at line 9918 @> WARNING failed to parse beta-factor at line 9919 @> WARNING failed to parse beta-factor at line 9920 @> WARNING failed to parse beta-factor at line 9921 @> WARNING failed to parse beta-factor at line 9922 @> WARNING failed to parse beta-factor at line 9923 @> WARNING failed to parse beta-factor at line 9924 @> WARNING failed to parse beta-factor at line 9925 @> WARNING failed to parse beta-factor at line 9926 @> WARNING failed to parse beta-factor at line 9927 @> WARNING failed to parse beta-factor at line 9928 @> WARNING failed to parse beta-factor at line 9929 @> WARNING failed to parse beta-factor at line 9930 @> WARNING failed to parse beta-factor at line 9931 @> WARNING failed to parse beta-factor at line 9932 @> WARNING failed to parse beta-factor at line 9933 @> WARNING failed to parse beta-factor at line 9934 @> WARNING failed to parse beta-factor at line 9935 @> WARNING failed to parse beta-factor at line 9936 @> WARNING failed to parse beta-factor at line 9937 @> WARNING failed to parse beta-factor at line 9938 @> WARNING failed to parse beta-factor at line 9939 @> WARNING failed to parse beta-factor at line 9940 @> WARNING failed to parse beta-factor at line 9941 @> WARNING failed to parse beta-factor at line 9942 @> WARNING failed to parse beta-factor at line 9943 @> WARNING failed to parse beta-factor at line 9944 @> WARNING failed to parse beta-factor at line 9945 @> WARNING failed to parse beta-factor at line 9946 @> WARNING failed to parse beta-factor at line 9947 @> WARNING failed to parse beta-factor at line 9948 @> WARNING failed to parse beta-factor at line 9949 @> WARNING failed to parse beta-factor at line 9950 @> WARNING failed to parse beta-factor at line 9951 @> WARNING failed to parse beta-factor at line 9952 @> WARNING failed to parse beta-factor at line 9953 @> WARNING failed to parse beta-factor at line 9954 @> WARNING failed to parse beta-factor at line 9955 @> WARNING failed to parse beta-factor at line 9956 @> WARNING failed to parse beta-factor at line 9957 @> WARNING failed to parse beta-factor at line 9958 @> WARNING failed to parse beta-factor at line 9959 @> WARNING failed to parse beta-factor at line 9960 @> WARNING failed to parse beta-factor at line 9961 @> WARNING failed to parse beta-factor at line 9962 @> WARNING failed to parse beta-factor at line 9963 @> WARNING failed to parse beta-factor at line 9964 @> WARNING failed to parse beta-factor at line 9965 @> WARNING failed to parse beta-factor at line 9966 @> WARNING failed to parse beta-factor at line 9967 @> WARNING failed to parse beta-factor at line 9968 @> WARNING failed to parse beta-factor at line 9969 @> WARNING failed to parse beta-factor at line 9970 @> WARNING failed to parse beta-factor at line 9971 @> WARNING failed to parse beta-factor at line 9972 @> WARNING failed to parse beta-factor at line 9973 @> WARNING failed to parse beta-factor at line 9974 @> WARNING failed to parse beta-factor at line 9975 @> WARNING failed to parse beta-factor at line 9976 @> WARNING failed to parse beta-factor at line 9977 @> WARNING failed to parse beta-factor at line 9978 @> WARNING failed to parse beta-factor at line 9979 @> WARNING failed to parse beta-factor at line 9980 @> WARNING failed to parse beta-factor at line 9981 @> WARNING failed to parse beta-factor at line 9982 @> WARNING failed to parse beta-factor at line 9983 @> WARNING failed to parse beta-factor at line 9984 @> WARNING failed to parse beta-factor at line 9985 @> WARNING failed to parse beta-factor at line 9986 @> WARNING failed to parse beta-factor at line 9987 @> WARNING failed to parse beta-factor at line 9988 @> WARNING failed to parse beta-factor at line 9989 @> WARNING failed to parse beta-factor at line 9990 @> WARNING failed to parse beta-factor at line 9991 @> WARNING failed to parse beta-factor at line 9992 @> WARNING failed to parse beta-factor at line 9993 @> WARNING failed to parse beta-factor at line 9994 @> WARNING failed to parse beta-factor at line 9995 @> WARNING failed to parse beta-factor at line 9996 @> WARNING failed to parse beta-factor at line 9997 @> WARNING failed to parse beta-factor at line 9998 @> WARNING failed to parse beta-factor at line 9999 @> WARNING failed to parse beta-factor at line 10000 @> WARNING failed to parse beta-factor at line 10001 @> WARNING failed to parse beta-factor at line 10002 @> WARNING failed to parse beta-factor at line 10003 @> WARNING failed to parse beta-factor at line 10004 @> WARNING failed to parse beta-factor at line 10005 @> WARNING failed to parse beta-factor at line 10006 @> WARNING failed to parse beta-factor at line 10007 @> WARNING failed to parse beta-factor at line 10008 @> WARNING failed to parse beta-factor at line 10009 @> WARNING failed to parse beta-factor at line 10010 @> WARNING failed to parse beta-factor at line 10011 @> WARNING failed to parse beta-factor at line 10012 @> WARNING failed to parse beta-factor at line 10013 @> WARNING failed to parse beta-factor at line 10014 @> WARNING failed to parse beta-factor at line 10015 @> WARNING failed to parse beta-factor at line 10016 @> WARNING failed to parse beta-factor at line 10017 @> WARNING failed to parse beta-factor at line 10018 @> WARNING failed to parse beta-factor at line 10019 @> WARNING failed to parse beta-factor at line 10020 @> WARNING failed to parse beta-factor at line 10021 @> WARNING failed to parse beta-factor at line 10022 @> WARNING failed to parse beta-factor at line 10023 @> WARNING failed to parse beta-factor at line 10024 @> WARNING failed to parse beta-factor at line 10025 @> WARNING failed to parse beta-factor at line 10026 @> WARNING failed to parse beta-factor at line 10027 @> WARNING failed to parse beta-factor at line 10028 @> WARNING failed to parse beta-factor at line 10029 @> WARNING failed to parse beta-factor at line 10030 @> WARNING failed to parse beta-factor at line 10031 @> WARNING failed to parse beta-factor at line 10032 @> WARNING failed to parse beta-factor at line 10033 @> WARNING failed to parse beta-factor at line 10034 @> WARNING failed to parse beta-factor at line 10035 @> WARNING failed to parse beta-factor at line 10036 @> WARNING failed to parse beta-factor at line 10037 @> WARNING failed to parse beta-factor at line 10038 @> WARNING failed to parse beta-factor at line 10039 @> WARNING failed to parse beta-factor at line 10040 @> WARNING failed to parse beta-factor at line 10041 @> WARNING failed to parse beta-factor at line 10042 @> WARNING failed to parse beta-factor at line 10043 @> WARNING failed to parse beta-factor at line 10044 @> WARNING failed to parse beta-factor at line 10045 @> WARNING failed to parse beta-factor at line 10046 @> WARNING failed to parse beta-factor at line 10047 @> WARNING failed to parse beta-factor at line 10048 @> WARNING failed to parse beta-factor at line 10049 @> WARNING failed to parse beta-factor at line 10050 @> WARNING failed to parse beta-factor at line 10051 @> WARNING failed to parse beta-factor at line 10052 @> WARNING failed to parse beta-factor at line 10053 @> WARNING failed to parse beta-factor at line 10054 @> WARNING failed to parse beta-factor at line 10055 @> WARNING failed to parse beta-factor at line 10056 @> WARNING failed to parse beta-factor at line 10057 @> WARNING failed to parse beta-factor at line 10058 @> WARNING failed to parse beta-factor at line 10059 @> WARNING failed to parse beta-factor at line 10060 @> WARNING failed to parse beta-factor at line 10061 @> WARNING failed to parse beta-factor at line 10062 @> WARNING failed to parse beta-factor at line 10063 @> WARNING failed to parse beta-factor at line 10064 @> WARNING failed to parse beta-factor at line 10065 @> WARNING failed to parse beta-factor at line 10066 @> WARNING failed to parse beta-factor at line 10067 @> WARNING failed to parse beta-factor at line 10068 @> WARNING failed to parse beta-factor at line 10069 @> WARNING failed to parse beta-factor at line 10070 @> WARNING failed to parse beta-factor at line 10071 @> WARNING failed to parse beta-factor at line 10072 @> WARNING failed to parse beta-factor at line 10073 @> WARNING failed to parse beta-factor at line 10074 @> WARNING failed to parse beta-factor at line 10075 @> WARNING failed to parse beta-factor at line 10076 @> WARNING failed to parse beta-factor at line 10077 @> WARNING failed to parse beta-factor at line 10078 @> WARNING failed to parse beta-factor at line 10079 @> WARNING failed to parse beta-factor at line 10080 @> WARNING failed to parse beta-factor at line 10081 @> WARNING failed to parse beta-factor at line 10082 @> WARNING failed to parse beta-factor at line 10083 @> WARNING failed to parse beta-factor at line 10084 @> WARNING failed to parse beta-factor at line 10085 @> WARNING failed to parse beta-factor at line 10086 @> WARNING failed to parse beta-factor at line 10087 @> WARNING failed to parse beta-factor at line 10088 @> WARNING failed to parse beta-factor at line 10089 @> WARNING failed to parse beta-factor at line 10090 @> WARNING failed to parse beta-factor at line 10091 @> WARNING failed to parse beta-factor at line 10092 @> WARNING failed to parse beta-factor at line 10093 @> WARNING failed to parse beta-factor at line 10094 @> WARNING failed to parse beta-factor at line 10095 @> WARNING failed to parse beta-factor at line 10096 @> WARNING failed to parse beta-factor at line 10097 @> WARNING failed to parse beta-factor at line 10098 @> WARNING failed to parse beta-factor at line 10099 @> WARNING failed to parse beta-factor at line 10100 @> WARNING failed to parse beta-factor at line 10101 @> WARNING failed to parse beta-factor at line 10102 @> WARNING failed to parse beta-factor at line 10103 @> WARNING failed to parse beta-factor at line 10104 @> WARNING failed to parse beta-factor at line 10105 @> WARNING failed to parse beta-factor at line 10106 @> WARNING failed to parse beta-factor at line 10107 @> WARNING failed to parse beta-factor at line 10108 @> WARNING failed to parse beta-factor at line 10109 @> WARNING failed to parse beta-factor at line 10110 @> WARNING failed to parse beta-factor at line 10111 @> WARNING failed to parse beta-factor at line 10112 @> WARNING failed to parse beta-factor at line 10113 @> WARNING failed to parse beta-factor at line 10114 @> WARNING failed to parse beta-factor at line 10115 @> WARNING failed to parse beta-factor at line 10116 @> WARNING failed to parse beta-factor at line 10117 @> WARNING failed to parse beta-factor at line 10118 @> WARNING failed to parse beta-factor at line 10119 @> WARNING failed to parse beta-factor at line 10120 @> WARNING failed to parse beta-factor at line 10121 @> WARNING failed to parse beta-factor at line 10122 @> WARNING failed to parse beta-factor at line 10123 @> WARNING failed to parse beta-factor at line 10124 @> WARNING failed to parse beta-factor at line 10125 @> WARNING failed to parse beta-factor at line 10126 @> WARNING failed to parse beta-factor at line 10127 @> WARNING failed to parse beta-factor at line 10128 @> WARNING failed to parse beta-factor at line 10129 @> WARNING failed to parse beta-factor at line 10130 @> WARNING failed to parse beta-factor at line 10131 @> WARNING failed to parse beta-factor at line 10132 @> WARNING failed to parse beta-factor at line 10133 @> WARNING failed to parse beta-factor at line 10134 @> WARNING failed to parse beta-factor at line 10135 @> WARNING failed to parse beta-factor at line 10136 @> WARNING failed to parse beta-factor at line 10137 @> WARNING failed to parse beta-factor at line 10138 @> WARNING failed to parse beta-factor at line 10139 @> WARNING failed to parse beta-factor at line 10140 @> WARNING failed to parse beta-factor at line 10141 @> WARNING failed to parse beta-factor at line 10142 @> WARNING failed to parse beta-factor at line 10143 @> WARNING failed to parse beta-factor at line 10144 @> WARNING failed to parse beta-factor at line 10145 @> WARNING failed to parse beta-factor at line 10146 @> WARNING failed to parse beta-factor at line 10147 @> WARNING failed to parse beta-factor at line 10148 @> WARNING failed to parse beta-factor at line 10149 @> WARNING failed to parse beta-factor at line 10150 @> WARNING failed to parse beta-factor at line 10151 @> WARNING failed to parse beta-factor at line 10152 @> WARNING failed to parse beta-factor at line 10153 @> WARNING failed to parse beta-factor at line 10154 @> WARNING failed to parse beta-factor at line 10155 @> WARNING failed to parse beta-factor at line 10156 @> WARNING failed to parse beta-factor at line 10157 @> WARNING failed to parse beta-factor at line 10158 @> WARNING failed to parse beta-factor at line 10159 @> WARNING failed to parse beta-factor at line 10160 @> WARNING failed to parse beta-factor at line 10161 @> WARNING failed to parse beta-factor at line 10162 @> WARNING failed to parse beta-factor at line 10163 @> WARNING failed to parse beta-factor at line 10164 @> WARNING failed to parse beta-factor at line 10165 @> WARNING failed to parse beta-factor at line 10166 @> WARNING failed to parse beta-factor at line 10167 @> WARNING failed to parse beta-factor at line 10168 @> WARNING failed to parse beta-factor at line 10169 @> WARNING failed to parse beta-factor at line 10170 @> WARNING failed to parse beta-factor at line 10171 @> WARNING failed to parse beta-factor at line 10172 @> WARNING failed to parse beta-factor at line 10173 @> WARNING failed to parse beta-factor at line 10174 @> WARNING failed to parse beta-factor at line 10175 @> WARNING failed to parse beta-factor at line 10176 @> WARNING failed to parse beta-factor at line 10177 @> WARNING failed to parse beta-factor at line 10178 @> WARNING failed to parse beta-factor at line 10179 @> WARNING failed to parse beta-factor at line 10180 @> WARNING failed to parse beta-factor at line 10181 @> WARNING failed to parse beta-factor at line 10182 @> WARNING failed to parse beta-factor at line 10183 @> WARNING failed to parse beta-factor at line 10184 @> WARNING failed to parse beta-factor at line 10185 @> WARNING failed to parse beta-factor at line 10186 @> WARNING failed to parse beta-factor at line 10187 @> WARNING failed to parse beta-factor at line 10188 @> WARNING failed to parse beta-factor at line 10189 @> WARNING failed to parse beta-factor at line 10190 @> WARNING failed to parse beta-factor at line 10191 @> WARNING failed to parse beta-factor at line 10192 @> WARNING failed to parse beta-factor at line 10193 @> WARNING failed to parse beta-factor at line 10194 @> WARNING failed to parse beta-factor at line 10195 @> WARNING failed to parse beta-factor at line 10196 @> WARNING failed to parse beta-factor at line 10197 @> WARNING failed to parse beta-factor at line 10198 @> WARNING failed to parse beta-factor at line 10199 @> WARNING failed to parse beta-factor at line 10200 @> WARNING failed to parse beta-factor at line 10201 @> WARNING failed to parse beta-factor at line 10202 @> WARNING failed to parse beta-factor at line 10203 @> WARNING failed to parse beta-factor at line 10204 @> WARNING failed to parse beta-factor at line 10205 @> WARNING failed to parse beta-factor at line 10206 @> WARNING failed to parse beta-factor at line 10207 @> WARNING failed to parse beta-factor at line 10208 @> WARNING failed to parse beta-factor at line 10209 @> WARNING failed to parse beta-factor at line 10210 @> WARNING failed to parse beta-factor at line 10211 @> WARNING failed to parse beta-factor at line 10212 @> WARNING failed to parse beta-factor at line 10213 @> WARNING failed to parse beta-factor at line 10214 @> WARNING failed to parse beta-factor at line 10215 @> WARNING failed to parse beta-factor at line 10216 @> WARNING failed to parse beta-factor at line 10217 @> WARNING failed to parse beta-factor at line 10218 @> WARNING failed to parse beta-factor at line 10219 @> WARNING failed to parse beta-factor at line 10220 @> WARNING failed to parse beta-factor at line 10221 @> WARNING failed to parse beta-factor at line 10222 @> WARNING failed to parse beta-factor at line 10223 @> WARNING failed to parse beta-factor at line 10224 @> WARNING failed to parse beta-factor at line 10225 @> WARNING failed to parse beta-factor at line 10226 @> WARNING failed to parse beta-factor at line 10227 @> WARNING failed to parse beta-factor at line 10228 @> WARNING failed to parse beta-factor at line 10229 @> WARNING failed to parse beta-factor at line 10230 @> WARNING failed to parse beta-factor at line 10231 @> WARNING failed to parse beta-factor at line 10232 @> WARNING failed to parse beta-factor at line 10233 @> WARNING failed to parse beta-factor at line 10234 @> WARNING failed to parse beta-factor at line 10235 @> WARNING failed to parse beta-factor at line 10236 @> WARNING failed to parse beta-factor at line 10237 @> WARNING failed to parse beta-factor at line 10238 @> WARNING failed to parse beta-factor at line 10239 @> WARNING failed to parse beta-factor at line 10240 @> WARNING failed to parse beta-factor at line 10241 @> WARNING failed to parse beta-factor at line 10242 @> WARNING failed to parse beta-factor at line 10243 @> WARNING failed to parse beta-factor at line 10244 @> WARNING failed to parse beta-factor at line 10245 @> WARNING failed to parse beta-factor at line 10246 @> WARNING failed to parse beta-factor at line 10247 @> WARNING failed to parse beta-factor at line 10248 @> WARNING failed to parse beta-factor at line 10249 @> WARNING failed to parse beta-factor at line 10250 @> WARNING failed to parse beta-factor at line 10251 @> WARNING failed to parse beta-factor at line 10252 @> WARNING failed to parse beta-factor at line 10253 @> WARNING failed to parse beta-factor at line 10254 @> WARNING failed to parse beta-factor at line 10255 @> WARNING failed to parse beta-factor at line 10256 @> WARNING failed to parse beta-factor at line 10257 @> WARNING failed to parse beta-factor at line 10258 @> WARNING failed to parse beta-factor at line 10259 @> WARNING failed to parse beta-factor at line 10260 @> WARNING failed to parse beta-factor at line 10261 @> WARNING failed to parse beta-factor at line 10262 @> WARNING failed to parse beta-factor at line 10263 @> WARNING failed to parse beta-factor at line 10264 @> WARNING failed to parse beta-factor at line 10265 @> WARNING failed to parse beta-factor at line 10266 @> WARNING failed to parse beta-factor at line 10267 @> WARNING failed to parse beta-factor at line 10268 @> WARNING failed to parse beta-factor at line 10269 @> WARNING failed to parse beta-factor at line 10270 @> WARNING failed to parse beta-factor at line 10271 @> WARNING failed to parse beta-factor at line 10272 @> WARNING failed to parse beta-factor at line 10273 @> WARNING failed to parse beta-factor at line 10274 @> WARNING failed to parse beta-factor at line 10275 @> WARNING failed to parse beta-factor at line 10276 @> WARNING failed to parse beta-factor at line 10277 @> WARNING failed to parse beta-factor at line 10278 @> WARNING failed to parse beta-factor at line 10279 @> WARNING failed to parse beta-factor at line 10280 @> WARNING failed to parse beta-factor at line 10281 @> WARNING failed to parse beta-factor at line 10282 @> WARNING failed to parse beta-factor at line 10283 @> WARNING failed to parse beta-factor at line 10284 @> WARNING failed to parse beta-factor at line 10285 @> WARNING failed to parse beta-factor at line 10286 @> WARNING failed to parse beta-factor at line 10287 @> WARNING failed to parse beta-factor at line 10288 @> WARNING failed to parse beta-factor at line 10289 @> WARNING failed to parse beta-factor at line 10290 @> WARNING failed to parse beta-factor at line 10291 @> WARNING failed to parse beta-factor at line 10292 @> WARNING failed to parse beta-factor at line 10293 @> WARNING failed to parse beta-factor at line 10294 @> WARNING failed to parse beta-factor at line 10295 @> WARNING failed to parse beta-factor at line 10296 @> WARNING failed to parse beta-factor at line 10297 @> WARNING failed to parse beta-factor at line 10298 @> WARNING failed to parse beta-factor at line 10299 @> WARNING failed to parse beta-factor at line 10300 @> WARNING failed to parse beta-factor at line 10301 @> WARNING failed to parse beta-factor at line 10302 @> WARNING failed to parse beta-factor at line 10303 @> WARNING failed to parse beta-factor at line 10304 @> WARNING failed to parse beta-factor at line 10305 @> WARNING failed to parse beta-factor at line 10306 @> WARNING failed to parse beta-factor at line 10307 @> WARNING failed to parse beta-factor at line 10308 @> WARNING failed to parse beta-factor at line 10309 @> WARNING failed to parse beta-factor at line 10310 @> WARNING failed to parse beta-factor at line 10311 @> WARNING failed to parse beta-factor at line 10312 @> WARNING failed to parse beta-factor at line 10313 @> WARNING failed to parse beta-factor at line 10314 @> WARNING failed to parse beta-factor at line 10315 @> WARNING failed to parse beta-factor at line 10316 @> WARNING failed to parse beta-factor at line 10317 @> WARNING failed to parse beta-factor at line 10318 @> WARNING failed to parse beta-factor at line 10319 @> WARNING failed to parse beta-factor at line 10320 @> WARNING failed to parse beta-factor at line 10321 @> WARNING failed to parse beta-factor at line 10322 @> WARNING failed to parse beta-factor at line 10323 @> WARNING failed to parse beta-factor at line 10324 @> WARNING failed to parse beta-factor at line 10325 @> WARNING failed to parse beta-factor at line 10326 @> WARNING failed to parse beta-factor at line 10327 @> WARNING failed to parse beta-factor at line 10328 @> WARNING failed to parse beta-factor at line 10329 @> WARNING failed to parse beta-factor at line 10330 @> WARNING failed to parse beta-factor at line 10331 @> WARNING failed to parse beta-factor at line 10332 @> WARNING failed to parse beta-factor at line 10333 @> WARNING failed to parse beta-factor at line 10334 @> WARNING failed to parse beta-factor at line 10335 @> WARNING failed to parse beta-factor at line 10336 @> WARNING failed to parse beta-factor at line 10337 @> WARNING failed to parse beta-factor at line 10338 @> WARNING failed to parse beta-factor at line 10339 @> WARNING failed to parse beta-factor at line 10340 @> WARNING failed to parse beta-factor at line 10341 @> WARNING failed to parse beta-factor at line 10342 @> WARNING failed to parse beta-factor at line 10343 @> WARNING failed to parse beta-factor at line 10344 @> WARNING failed to parse beta-factor at line 10345 @> WARNING failed to parse beta-factor at line 10346 @> WARNING failed to parse beta-factor at line 10347 @> WARNING failed to parse beta-factor at line 10348 @> WARNING failed to parse beta-factor at line 10349 @> WARNING failed to parse beta-factor at line 10350 @> WARNING failed to parse beta-factor at line 10351 @> WARNING failed to parse beta-factor at line 10352 @> WARNING failed to parse beta-factor at line 10353 @> WARNING failed to parse beta-factor at line 10354 @> WARNING failed to parse beta-factor at line 10355 @> WARNING failed to parse beta-factor at line 10356 @> WARNING failed to parse beta-factor at line 10357 @> WARNING failed to parse beta-factor at line 10358 @> WARNING failed to parse beta-factor at line 10359 @> WARNING failed to parse beta-factor at line 10360 @> WARNING failed to parse beta-factor at line 10361 @> WARNING failed to parse beta-factor at line 10362 @> WARNING failed to parse beta-factor at line 10363 @> WARNING failed to parse beta-factor at line 10364 @> WARNING failed to parse beta-factor at line 10365 @> WARNING failed to parse beta-factor at line 10366 @> WARNING failed to parse beta-factor at line 10367 @> WARNING failed to parse beta-factor at line 10368 @> WARNING failed to parse beta-factor at line 10369 @> WARNING failed to parse beta-factor at line 10370 @> WARNING failed to parse beta-factor at line 10371 @> WARNING failed to parse beta-factor at line 10372 @> WARNING failed to parse beta-factor at line 10373 @> WARNING failed to parse beta-factor at line 10374 @> WARNING failed to parse beta-factor at line 10375 @> WARNING failed to parse beta-factor at line 10376 @> WARNING failed to parse beta-factor at line 10377 @> WARNING failed to parse beta-factor at line 10378 @> WARNING failed to parse beta-factor at line 10379 @> WARNING failed to parse beta-factor at line 10380 @> WARNING failed to parse beta-factor at line 10381 @> WARNING failed to parse beta-factor at line 10382 @> WARNING failed to parse beta-factor at line 10383 @> WARNING failed to parse beta-factor at line 10384 @> WARNING failed to parse beta-factor at line 10385 @> WARNING failed to parse beta-factor at line 10386 @> WARNING failed to parse beta-factor at line 10387 @> WARNING failed to parse beta-factor at line 10388 @> WARNING failed to parse beta-factor at line 10389 @> WARNING failed to parse beta-factor at line 10390 @> WARNING failed to parse beta-factor at line 10391 @> WARNING failed to parse beta-factor at line 10392 @> WARNING failed to parse beta-factor at line 10393 @> WARNING failed to parse beta-factor at line 10394 @> WARNING failed to parse beta-factor at line 10395 @> WARNING failed to parse beta-factor at line 10396 @> WARNING failed to parse beta-factor at line 10397 @> WARNING failed to parse beta-factor at line 10398 @> WARNING failed to parse beta-factor at line 10399 @> WARNING failed to parse beta-factor at line 10400 @> WARNING failed to parse beta-factor at line 10401 @> WARNING failed to parse beta-factor at line 10402 @> WARNING failed to parse beta-factor at line 10403 @> WARNING failed to parse beta-factor at line 10404 @> WARNING failed to parse beta-factor at line 10405 @> WARNING failed to parse beta-factor at line 10406 @> WARNING failed to parse beta-factor at line 10407 @> WARNING failed to parse beta-factor at line 10408 @> WARNING failed to parse beta-factor at line 10409 @> WARNING failed to parse beta-factor at line 10410 @> WARNING failed to parse beta-factor at line 10411 @> WARNING failed to parse beta-factor at line 10412 @> WARNING failed to parse beta-factor at line 10413 @> WARNING failed to parse beta-factor at line 10414 @> WARNING failed to parse beta-factor at line 10415 @> WARNING failed to parse beta-factor at line 10416 @> WARNING failed to parse beta-factor at line 10417 @> WARNING failed to parse beta-factor at line 10418 @> WARNING failed to parse beta-factor at line 10419 @> WARNING failed to parse beta-factor at line 10420 @> WARNING failed to parse beta-factor at line 10421 @> WARNING failed to parse beta-factor at line 10422 @> WARNING failed to parse beta-factor at line 10423 @> WARNING failed to parse beta-factor at line 10424 @> WARNING failed to parse beta-factor at line 10425 @> WARNING failed to parse beta-factor at line 10426 @> WARNING failed to parse beta-factor at line 10427 @> WARNING failed to parse beta-factor at line 10428 @> WARNING failed to parse beta-factor at line 10429 @> WARNING failed to parse beta-factor at line 10430 @> WARNING failed to parse beta-factor at line 10431 @> WARNING failed to parse beta-factor at line 10432 @> WARNING failed to parse beta-factor at line 10433 @> WARNING failed to parse beta-factor at line 10434 @> WARNING failed to parse beta-factor at line 10435 @> WARNING failed to parse beta-factor at line 10436 @> WARNING failed to parse beta-factor at line 10437 @> WARNING failed to parse beta-factor at line 10438 @> WARNING failed to parse beta-factor at line 10439 @> WARNING failed to parse beta-factor at line 10440 @> WARNING failed to parse beta-factor at line 10441 @> WARNING failed to parse beta-factor at line 10442 @> WARNING failed to parse beta-factor at line 10443 @> WARNING failed to parse beta-factor at line 10444 @> WARNING failed to parse beta-factor at line 10445 @> WARNING failed to parse beta-factor at line 10446 @> WARNING failed to parse beta-factor at line 10447 @> WARNING failed to parse beta-factor at line 10448 @> WARNING failed to parse beta-factor at line 10449 @> WARNING failed to parse beta-factor at line 10450 @> WARNING failed to parse beta-factor at line 10451 @> WARNING failed to parse beta-factor at line 10452 @> WARNING failed to parse beta-factor at line 10453 @> WARNING failed to parse beta-factor at line 10454 @> WARNING failed to parse beta-factor at line 10455 @> WARNING failed to parse beta-factor at line 10456 @> WARNING failed to parse beta-factor at line 10457 @> WARNING failed to parse beta-factor at line 10458 @> WARNING failed to parse beta-factor at line 10459 @> WARNING failed to parse beta-factor at line 10460 @> WARNING failed to parse beta-factor at line 10461 @> WARNING failed to parse beta-factor at line 10462 @> WARNING failed to parse beta-factor at line 10463 @> WARNING failed to parse beta-factor at line 10464 @> WARNING failed to parse beta-factor at line 10465 @> WARNING failed to parse beta-factor at line 10466 @> WARNING failed to parse beta-factor at line 10467 @> WARNING failed to parse beta-factor at line 10468 @> WARNING failed to parse beta-factor at line 10469 @> WARNING failed to parse beta-factor at line 10470 @> WARNING failed to parse beta-factor at line 10471 @> WARNING failed to parse beta-factor at line 10472 @> WARNING failed to parse beta-factor at line 10473 @> WARNING failed to parse beta-factor at line 10474 @> WARNING failed to parse beta-factor at line 10475 @> WARNING failed to parse beta-factor at line 10476 @> WARNING failed to parse beta-factor at line 10477 @> WARNING failed to parse beta-factor at line 10478 @> WARNING failed to parse beta-factor at line 10479 @> WARNING failed to parse beta-factor at line 10480 @> WARNING failed to parse beta-factor at line 10481 @> WARNING failed to parse beta-factor at line 10482 @> WARNING failed to parse beta-factor at line 10483 @> WARNING failed to parse beta-factor at line 10484 @> WARNING failed to parse beta-factor at line 10485 @> WARNING failed to parse beta-factor at line 10486 @> WARNING failed to parse beta-factor at line 10487 @> WARNING failed to parse beta-factor at line 10488 @> WARNING failed to parse beta-factor at line 10489 @> WARNING failed to parse beta-factor at line 10490 @> WARNING failed to parse beta-factor at line 10491 @> WARNING failed to parse beta-factor at line 10492 @> WARNING failed to parse beta-factor at line 10493 @> WARNING failed to parse beta-factor at line 10494 @> WARNING failed to parse beta-factor at line 10495 @> WARNING failed to parse beta-factor at line 10496 @> WARNING failed to parse beta-factor at line 10497 @> WARNING failed to parse beta-factor at line 10498 @> WARNING failed to parse beta-factor at line 10499 @> WARNING failed to parse beta-factor at line 10500 @> WARNING failed to parse beta-factor at line 10501 @> WARNING failed to parse beta-factor at line 10502 @> WARNING failed to parse beta-factor at line 10503 @> WARNING failed to parse beta-factor at line 10504 @> WARNING failed to parse beta-factor at line 10505 @> WARNING failed to parse beta-factor at line 10506 @> WARNING failed to parse beta-factor at line 10507 @> WARNING failed to parse beta-factor at line 10508 @> WARNING failed to parse beta-factor at line 10509 @> WARNING failed to parse beta-factor at line 10510 @> WARNING failed to parse beta-factor at line 10511 @> WARNING failed to parse beta-factor at line 10512 @> WARNING failed to parse beta-factor at line 10513 @> WARNING failed to parse beta-factor at line 10514 @> WARNING failed to parse beta-factor at line 10515 @> WARNING failed to parse beta-factor at line 10516 @> WARNING failed to parse beta-factor at line 10517 @> WARNING failed to parse beta-factor at line 10518 @> WARNING failed to parse beta-factor at line 10519 @> WARNING failed to parse beta-factor at line 10520 @> WARNING failed to parse beta-factor at line 10521 @> WARNING failed to parse beta-factor at line 10522 @> WARNING failed to parse beta-factor at line 10523 @> WARNING failed to parse beta-factor at line 10524 @> WARNING failed to parse beta-factor at line 10525 @> WARNING failed to parse beta-factor at line 10526 @> WARNING failed to parse beta-factor at line 10527 @> WARNING failed to parse beta-factor at line 10528 @> WARNING failed to parse beta-factor at line 10529 @> WARNING failed to parse beta-factor at line 10530 @> WARNING failed to parse beta-factor at line 10531 @> WARNING failed to parse beta-factor at line 10532 @> WARNING failed to parse beta-factor at line 10533 @> WARNING failed to parse beta-factor at line 10534 @> WARNING failed to parse beta-factor at line 10535 @> WARNING failed to parse beta-factor at line 10536 @> WARNING failed to parse beta-factor at line 10537 @> WARNING failed to parse beta-factor at line 10538 @> WARNING failed to parse beta-factor at line 10539 @> WARNING failed to parse beta-factor at line 10540 @> WARNING failed to parse beta-factor at line 10541 @> WARNING failed to parse beta-factor at line 10542 @> WARNING failed to parse beta-factor at line 10543 @> WARNING failed to parse beta-factor at line 10544 @> WARNING failed to parse beta-factor at line 10545 @> WARNING failed to parse beta-factor at line 10546 @> WARNING failed to parse beta-factor at line 10547 @> WARNING failed to parse beta-factor at line 10548 @> WARNING failed to parse beta-factor at line 10549 @> WARNING failed to parse beta-factor at line 10550 @> WARNING failed to parse beta-factor at line 10551 @> WARNING failed to parse beta-factor at line 10552 @> WARNING failed to parse beta-factor at line 10553 @> WARNING failed to parse beta-factor at line 10554 @> WARNING failed to parse beta-factor at line 10555 @> WARNING failed to parse beta-factor at line 10556 @> WARNING failed to parse beta-factor at line 10557 @> WARNING failed to parse beta-factor at line 10558 @> WARNING failed to parse beta-factor at line 10559 @> WARNING failed to parse beta-factor at line 10560 @> WARNING failed to parse beta-factor at line 10561 @> WARNING failed to parse beta-factor at line 10562 @> WARNING failed to parse beta-factor at line 10563 @> WARNING failed to parse beta-factor at line 10564 @> WARNING failed to parse beta-factor at line 10565 @> WARNING failed to parse beta-factor at line 10566 @> WARNING failed to parse beta-factor at line 10567 @> WARNING failed to parse beta-factor at line 10568 @> WARNING failed to parse beta-factor at line 10569 @> WARNING failed to parse beta-factor at line 10570 @> WARNING failed to parse beta-factor at line 10571 @> WARNING failed to parse beta-factor at line 10572 @> WARNING failed to parse beta-factor at line 10573 @> WARNING failed to parse beta-factor at line 10574 @> WARNING failed to parse beta-factor at line 10575 @> WARNING failed to parse beta-factor at line 10576 @> WARNING failed to parse beta-factor at line 10577 @> WARNING failed to parse beta-factor at line 10578 @> WARNING failed to parse beta-factor at line 10579 @> WARNING failed to parse beta-factor at line 10580 @> WARNING failed to parse beta-factor at line 10581 @> WARNING failed to parse beta-factor at line 10582 @> WARNING failed to parse beta-factor at line 10583 @> WARNING failed to parse beta-factor at line 10584 @> WARNING failed to parse beta-factor at line 10585 @> WARNING failed to parse beta-factor at line 10586 @> WARNING failed to parse beta-factor at line 10587 @> WARNING failed to parse beta-factor at line 10588 @> WARNING failed to parse beta-factor at line 10589 @> WARNING failed to parse beta-factor at line 10590 @> WARNING failed to parse beta-factor at line 10591 @> WARNING failed to parse beta-factor at line 10592 @> WARNING failed to parse beta-factor at line 10593 @> WARNING failed to parse beta-factor at line 10594 @> WARNING failed to parse beta-factor at line 10595 @> WARNING failed to parse beta-factor at line 10596 @> WARNING failed to parse beta-factor at line 10597 @> WARNING failed to parse beta-factor at line 10598 @> WARNING failed to parse beta-factor at line 10599 @> WARNING failed to parse beta-factor at line 10600 @> WARNING failed to parse beta-factor at line 10601 @> WARNING failed to parse beta-factor at line 10602 @> WARNING failed to parse beta-factor at line 10603 @> WARNING failed to parse beta-factor at line 10604 @> WARNING failed to parse beta-factor at line 10605 @> WARNING failed to parse beta-factor at line 10606 @> WARNING failed to parse beta-factor at line 10607 @> WARNING failed to parse beta-factor at line 10608 @> WARNING failed to parse beta-factor at line 10609 @> WARNING failed to parse beta-factor at line 10610 @> WARNING failed to parse beta-factor at line 10611 @> WARNING failed to parse beta-factor at line 10612 @> WARNING failed to parse beta-factor at line 10613 @> WARNING failed to parse beta-factor at line 10614 @> WARNING failed to parse beta-factor at line 10615 @> WARNING failed to parse beta-factor at line 10616 @> WARNING failed to parse beta-factor at line 10617 @> WARNING failed to parse beta-factor at line 10618 @> WARNING failed to parse beta-factor at line 10619 @> WARNING failed to parse beta-factor at line 10620 @> WARNING failed to parse beta-factor at line 10621 @> WARNING failed to parse beta-factor at line 10622 @> WARNING failed to parse beta-factor at line 10623 @> WARNING failed to parse beta-factor at line 10624 @> WARNING failed to parse beta-factor at line 10625 @> WARNING failed to parse beta-factor at line 10626 @> WARNING failed to parse beta-factor at line 10627 @> WARNING failed to parse beta-factor at line 10628 @> WARNING failed to parse beta-factor at line 10629 @> WARNING failed to parse beta-factor at line 10630 @> WARNING failed to parse beta-factor at line 10631 @> WARNING failed to parse beta-factor at line 10632 @> WARNING failed to parse beta-factor at line 10633 @> WARNING failed to parse beta-factor at line 10634 @> WARNING failed to parse beta-factor at line 10635 @> WARNING failed to parse beta-factor at line 10636 @> WARNING failed to parse beta-factor at line 10637 @> WARNING failed to parse beta-factor at line 10638 @> WARNING failed to parse beta-factor at line 10639 @> WARNING failed to parse beta-factor at line 10640 @> WARNING failed to parse beta-factor at line 10641 @> WARNING failed to parse beta-factor at line 10642 @> WARNING failed to parse beta-factor at line 10643 @> WARNING failed to parse beta-factor at line 10644 @> WARNING failed to parse beta-factor at line 10645 @> WARNING failed to parse beta-factor at line 10646 @> WARNING failed to parse beta-factor at line 10647 @> WARNING failed to parse beta-factor at line 10648 @> WARNING failed to parse beta-factor at line 10649 @> WARNING failed to parse beta-factor at line 10650 @> WARNING failed to parse beta-factor at line 10651 @> WARNING failed to parse beta-factor at line 10652 @> WARNING failed to parse beta-factor at line 10653 @> WARNING failed to parse beta-factor at line 10654 @> WARNING failed to parse beta-factor at line 10655 @> WARNING failed to parse beta-factor at line 10656 @> WARNING failed to parse beta-factor at line 10657 @> WARNING failed to parse beta-factor at line 10658 @> WARNING failed to parse beta-factor at line 10659 @> WARNING failed to parse beta-factor at line 10660 @> WARNING failed to parse beta-factor at line 10661 @> WARNING failed to parse beta-factor at line 10662 @> WARNING failed to parse beta-factor at line 10663 @> WARNING failed to parse beta-factor at line 10664 @> WARNING failed to parse beta-factor at line 10665 @> WARNING failed to parse beta-factor at line 10666 @> WARNING failed to parse beta-factor at line 10667 @> WARNING failed to parse beta-factor at line 10668 @> WARNING failed to parse beta-factor at line 10669 @> WARNING failed to parse beta-factor at line 10670 @> WARNING failed to parse beta-factor at line 10671 @> WARNING failed to parse beta-factor at line 10672 @> WARNING failed to parse beta-factor at line 10673 @> WARNING failed to parse beta-factor at line 10674 @> WARNING failed to parse beta-factor at line 10675 @> WARNING failed to parse beta-factor at line 10676 @> WARNING failed to parse beta-factor at line 10677 @> WARNING failed to parse beta-factor at line 10678 @> WARNING failed to parse beta-factor at line 10679 @> WARNING failed to parse beta-factor at line 10680 @> WARNING failed to parse beta-factor at line 10681 @> WARNING failed to parse beta-factor at line 10682 @> WARNING failed to parse beta-factor at line 10683 @> WARNING failed to parse beta-factor at line 10684 @> WARNING failed to parse beta-factor at line 10685 @> WARNING failed to parse beta-factor at line 10686 @> WARNING failed to parse beta-factor at line 10687 @> WARNING failed to parse beta-factor at line 10688 @> WARNING failed to parse beta-factor at line 10689 @> WARNING failed to parse beta-factor at line 10690 @> WARNING failed to parse beta-factor at line 10691 @> WARNING failed to parse beta-factor at line 10692 @> WARNING failed to parse beta-factor at line 10693 @> WARNING failed to parse beta-factor at line 10694 @> WARNING failed to parse beta-factor at line 10695 @> WARNING failed to parse beta-factor at line 10696 @> WARNING failed to parse beta-factor at line 10697 @> WARNING failed to parse beta-factor at line 10698 @> WARNING failed to parse beta-factor at line 10699 @> WARNING failed to parse beta-factor at line 10700 @> WARNING failed to parse beta-factor at line 10701 @> WARNING failed to parse beta-factor at line 10702 @> WARNING failed to parse beta-factor at line 10703 @> WARNING failed to parse beta-factor at line 10704 @> WARNING failed to parse beta-factor at line 10705 @> WARNING failed to parse beta-factor at line 10706 @> WARNING failed to parse beta-factor at line 10707 @> WARNING failed to parse beta-factor at line 10708 @> WARNING failed to parse beta-factor at line 10709 @> WARNING failed to parse beta-factor at line 10710 @> WARNING failed to parse beta-factor at line 10711 @> WARNING failed to parse beta-factor at line 10712 @> WARNING failed to parse beta-factor at line 10713 @> WARNING failed to parse beta-factor at line 10714 @> WARNING failed to parse beta-factor at line 10715 @> WARNING failed to parse beta-factor at line 10716 @> WARNING failed to parse beta-factor at line 10717 @> WARNING failed to parse beta-factor at line 10718 @> WARNING failed to parse beta-factor at line 10719 @> WARNING failed to parse beta-factor at line 10720 @> WARNING failed to parse beta-factor at line 10721 @> WARNING failed to parse beta-factor at line 10722 @> WARNING failed to parse beta-factor at line 10723 @> WARNING failed to parse beta-factor at line 10724 @> WARNING failed to parse beta-factor at line 10725 @> WARNING failed to parse beta-factor at line 10726 @> WARNING failed to parse beta-factor at line 10727 @> WARNING failed to parse beta-factor at line 10728 @> WARNING failed to parse beta-factor at line 10729 @> WARNING failed to parse beta-factor at line 10730 @> WARNING failed to parse beta-factor at line 10731 @> WARNING failed to parse beta-factor at line 10732 @> WARNING failed to parse beta-factor at line 10733 @> WARNING failed to parse beta-factor at line 10734 @> WARNING failed to parse beta-factor at line 10735 @> WARNING failed to parse beta-factor at line 10736 @> WARNING failed to parse beta-factor at line 10737 @> WARNING failed to parse beta-factor at line 10738 @> WARNING failed to parse beta-factor at line 10739 @> WARNING failed to parse beta-factor at line 10740 @> WARNING failed to parse beta-factor at line 10741 @> WARNING failed to parse beta-factor at line 10742 @> WARNING failed to parse beta-factor at line 10743 @> WARNING failed to parse beta-factor at line 10744 @> WARNING failed to parse beta-factor at line 10745 @> WARNING failed to parse beta-factor at line 10746 @> WARNING failed to parse beta-factor at line 10747 @> WARNING failed to parse beta-factor at line 10748 @> WARNING failed to parse beta-factor at line 10749 @> WARNING failed to parse beta-factor at line 10750 @> WARNING failed to parse beta-factor at line 10751 @> WARNING failed to parse beta-factor at line 10752 @> WARNING failed to parse beta-factor at line 10753 @> WARNING failed to parse beta-factor at line 10754 @> WARNING failed to parse beta-factor at line 10755 @> WARNING failed to parse beta-factor at line 10756 @> WARNING failed to parse beta-factor at line 10757 @> WARNING failed to parse beta-factor at line 10758 @> WARNING failed to parse beta-factor at line 10759 @> WARNING failed to parse beta-factor at line 10760 @> WARNING failed to parse beta-factor at line 10761 @> WARNING failed to parse beta-factor at line 10762 @> WARNING failed to parse beta-factor at line 10763 @> WARNING failed to parse beta-factor at line 10764 @> WARNING failed to parse beta-factor at line 10765 @> WARNING failed to parse beta-factor at line 10766 @> WARNING failed to parse beta-factor at line 10767 @> WARNING failed to parse beta-factor at line 10768 @> WARNING failed to parse beta-factor at line 10769 @> WARNING failed to parse beta-factor at line 10770 @> WARNING failed to parse beta-factor at line 10771 @> WARNING failed to parse beta-factor at line 10772 @> WARNING failed to parse beta-factor at line 10773 @> WARNING failed to parse beta-factor at line 10774 @> WARNING failed to parse beta-factor at line 10775 @> WARNING failed to parse beta-factor at line 10776 @> WARNING failed to parse beta-factor at line 10777 @> WARNING failed to parse beta-factor at line 10778 @> WARNING failed to parse beta-factor at line 10779 @> WARNING failed to parse beta-factor at line 10780 @> WARNING failed to parse beta-factor at line 10781 @> WARNING failed to parse beta-factor at line 10782 @> WARNING failed to parse beta-factor at line 10783 @> WARNING failed to parse beta-factor at line 10784 @> WARNING failed to parse beta-factor at line 10785 @> WARNING failed to parse beta-factor at line 10786 @> WARNING failed to parse beta-factor at line 10787 @> WARNING failed to parse beta-factor at line 10788 @> WARNING failed to parse beta-factor at line 10789 @> WARNING failed to parse beta-factor at line 10790 @> WARNING failed to parse beta-factor at line 10791 @> WARNING failed to parse beta-factor at line 10792 @> WARNING failed to parse beta-factor at line 10793 @> WARNING failed to parse beta-factor at line 10794 @> WARNING failed to parse beta-factor at line 10795 @> WARNING failed to parse beta-factor at line 10796 @> WARNING failed to parse beta-factor at line 10797 @> WARNING failed to parse beta-factor at line 10798 @> WARNING failed to parse beta-factor at line 10799 @> WARNING failed to parse beta-factor at line 10800 @> WARNING failed to parse beta-factor at line 10801 @> WARNING failed to parse beta-factor at line 10802 @> WARNING failed to parse beta-factor at line 10803 @> WARNING failed to parse beta-factor at line 10804 @> WARNING failed to parse beta-factor at line 10805 @> WARNING failed to parse beta-factor at line 10806 @> WARNING failed to parse beta-factor at line 10807 @> WARNING failed to parse beta-factor at line 10808 @> WARNING failed to parse beta-factor at line 10809 @> WARNING failed to parse beta-factor at line 10810 @> WARNING failed to parse beta-factor at line 10811 @> WARNING failed to parse beta-factor at line 10812 @> WARNING failed to parse beta-factor at line 10813 @> WARNING failed to parse beta-factor at line 10814 @> WARNING failed to parse beta-factor at line 10815 @> WARNING failed to parse beta-factor at line 10816 @> WARNING failed to parse beta-factor at line 10817 @> WARNING failed to parse beta-factor at line 10818 @> WARNING failed to parse beta-factor at line 10819 @> WARNING failed to parse beta-factor at line 10820 @> WARNING failed to parse beta-factor at line 10821 @> WARNING failed to parse beta-factor at line 10822 @> WARNING failed to parse beta-factor at line 10823 @> WARNING failed to parse beta-factor at line 10824 @> WARNING failed to parse beta-factor at line 10825 @> WARNING failed to parse beta-factor at line 10826 @> WARNING failed to parse beta-factor at line 10827 @> WARNING failed to parse beta-factor at line 10828 @> WARNING failed to parse beta-factor at line 10829 @> WARNING failed to parse beta-factor at line 10830 @> WARNING failed to parse beta-factor at line 10831 @> WARNING failed to parse beta-factor at line 10832 @> WARNING failed to parse beta-factor at line 10833 @> WARNING failed to parse beta-factor at line 10834 @> WARNING failed to parse beta-factor at line 10835 @> WARNING failed to parse beta-factor at line 10836 @> WARNING failed to parse beta-factor at line 10837 @> WARNING failed to parse beta-factor at line 10838 @> WARNING failed to parse beta-factor at line 10839 @> WARNING failed to parse beta-factor at line 10840 @> WARNING failed to parse beta-factor at line 10841 @> WARNING failed to parse beta-factor at line 10842 @> WARNING failed to parse beta-factor at line 10843 @> WARNING failed to parse beta-factor at line 10844 @> WARNING failed to parse beta-factor at line 10845 @> WARNING failed to parse beta-factor at line 10846 @> WARNING failed to parse beta-factor at line 10847 @> WARNING failed to parse beta-factor at line 10848 @> WARNING failed to parse beta-factor at line 10849 @> WARNING failed to parse beta-factor at line 10850 @> WARNING failed to parse beta-factor at line 10851 @> WARNING failed to parse beta-factor at line 10852 @> WARNING failed to parse beta-factor at line 10853 @> WARNING failed to parse beta-factor at line 10854 @> WARNING failed to parse beta-factor at line 10855 @> WARNING failed to parse beta-factor at line 10856 @> WARNING failed to parse beta-factor at line 10857 @> WARNING failed to parse beta-factor at line 10858 @> WARNING failed to parse beta-factor at line 10859 @> WARNING failed to parse beta-factor at line 10860 @> WARNING failed to parse beta-factor at line 10861 @> WARNING failed to parse beta-factor at line 10862 @> WARNING failed to parse beta-factor at line 10863 @> WARNING failed to parse beta-factor at line 10864 @> WARNING failed to parse beta-factor at line 10865 @> WARNING failed to parse beta-factor at line 10866 @> WARNING failed to parse beta-factor at line 10867 @> WARNING failed to parse beta-factor at line 10868 @> WARNING failed to parse beta-factor at line 10869 @> WARNING failed to parse beta-factor at line 10870 @> WARNING failed to parse beta-factor at line 10871 @> WARNING failed to parse beta-factor at line 10872 @> WARNING failed to parse beta-factor at line 10873 @> WARNING failed to parse beta-factor at line 10874 @> WARNING failed to parse beta-factor at line 10875 @> WARNING failed to parse beta-factor at line 10876 @> WARNING failed to parse beta-factor at line 10877 @> WARNING failed to parse beta-factor at line 10878 @> WARNING failed to parse beta-factor at line 10879 @> WARNING failed to parse beta-factor at line 10880 @> WARNING failed to parse beta-factor at line 10881 @> WARNING failed to parse beta-factor at line 10882 @> WARNING failed to parse beta-factor at line 10883 @> WARNING failed to parse beta-factor at line 10884 @> WARNING failed to parse beta-factor at line 10885 @> WARNING failed to parse beta-factor at line 10886 @> WARNING failed to parse beta-factor at line 10887 @> WARNING failed to parse beta-factor at line 10888 @> WARNING failed to parse beta-factor at line 10889 @> WARNING failed to parse beta-factor at line 10890 @> WARNING failed to parse beta-factor at line 10891 @> WARNING failed to parse beta-factor at line 10892 @> WARNING failed to parse beta-factor at line 10893 @> WARNING failed to parse beta-factor at line 10894 @> WARNING failed to parse beta-factor at line 10895 @> WARNING failed to parse beta-factor at line 10896 @> WARNING failed to parse beta-factor at line 10897 @> WARNING failed to parse beta-factor at line 10898 @> WARNING failed to parse beta-factor at line 10899 @> WARNING failed to parse beta-factor at line 10900 @> WARNING failed to parse beta-factor at line 10901 @> WARNING failed to parse beta-factor at line 10902 @> WARNING failed to parse beta-factor at line 10903 @> WARNING failed to parse beta-factor at line 10904 @> WARNING failed to parse beta-factor at line 10905 @> WARNING failed to parse beta-factor at line 10906 @> WARNING failed to parse beta-factor at line 10907 @> WARNING failed to parse beta-factor at line 10908 @> WARNING failed to parse beta-factor at line 10909 @> WARNING failed to parse beta-factor at line 10910 @> WARNING failed to parse beta-factor at line 10911 @> WARNING failed to parse beta-factor at line 10912 @> WARNING failed to parse beta-factor at line 10913 @> WARNING failed to parse beta-factor at line 10914 @> WARNING failed to parse beta-factor at line 10915 @> WARNING failed to parse beta-factor at line 10916 @> WARNING failed to parse beta-factor at line 10917 @> WARNING failed to parse beta-factor at line 10918 @> WARNING failed to parse beta-factor at line 10919 @> WARNING failed to parse beta-factor at line 10920 @> WARNING failed to parse beta-factor at line 10921 @> WARNING failed to parse beta-factor at line 10922 @> WARNING failed to parse beta-factor at line 10923 @> WARNING failed to parse beta-factor at line 10924 @> WARNING failed to parse beta-factor at line 10925 @> WARNING failed to parse beta-factor at line 10926 @> WARNING failed to parse beta-factor at line 10927 @> WARNING failed to parse beta-factor at line 10928 @> WARNING failed to parse beta-factor at line 10929 @> WARNING failed to parse beta-factor at line 10930 @> WARNING failed to parse beta-factor at line 10931 @> WARNING failed to parse beta-factor at line 10932 @> WARNING failed to parse beta-factor at line 10933 @> WARNING failed to parse beta-factor at line 10934 @> WARNING failed to parse beta-factor at line 10935 @> WARNING failed to parse beta-factor at line 10936 @> WARNING failed to parse beta-factor at line 10937 @> WARNING failed to parse beta-factor at line 10938 @> WARNING failed to parse beta-factor at line 10939 @> WARNING failed to parse beta-factor at line 10940 @> WARNING failed to parse beta-factor at line 10941 @> WARNING failed to parse beta-factor at line 10942 @> WARNING failed to parse beta-factor at line 10943 @> WARNING failed to parse beta-factor at line 10944 @> WARNING failed to parse beta-factor at line 10945 @> WARNING failed to parse beta-factor at line 10946 @> WARNING failed to parse beta-factor at line 10947 @> WARNING failed to parse beta-factor at line 10948 @> WARNING failed to parse beta-factor at line 10949 @> WARNING failed to parse beta-factor at line 10950 @> WARNING failed to parse beta-factor at line 10951 @> WARNING failed to parse beta-factor at line 10952 @> WARNING failed to parse beta-factor at line 10953 @> WARNING failed to parse beta-factor at line 10954 @> WARNING failed to parse beta-factor at line 10955 @> WARNING failed to parse beta-factor at line 10956 @> WARNING failed to parse beta-factor at line 10957 @> WARNING failed to parse beta-factor at line 10958 @> WARNING failed to parse beta-factor at line 10959 @> WARNING failed to parse beta-factor at line 10960 @> WARNING failed to parse beta-factor at line 10961 @> WARNING failed to parse beta-factor at line 10962 @> WARNING failed to parse beta-factor at line 10963 @> WARNING failed to parse beta-factor at line 10964 @> WARNING failed to parse beta-factor at line 10965 @> WARNING failed to parse beta-factor at line 10966 @> WARNING failed to parse beta-factor at line 10967 @> WARNING failed to parse beta-factor at line 10968 @> WARNING failed to parse beta-factor at line 10969 @> WARNING failed to parse beta-factor at line 10970 @> WARNING failed to parse beta-factor at line 10971 @> WARNING failed to parse beta-factor at line 10972 @> WARNING failed to parse beta-factor at line 10973 @> WARNING failed to parse beta-factor at line 10974 @> WARNING failed to parse beta-factor at line 10975 @> WARNING failed to parse beta-factor at line 10976 @> WARNING failed to parse beta-factor at line 10977 @> WARNING failed to parse beta-factor at line 10978 @> WARNING failed to parse beta-factor at line 10979 @> WARNING failed to parse beta-factor at line 10980 @> WARNING failed to parse beta-factor at line 10981 @> WARNING failed to parse beta-factor at line 10982 @> WARNING failed to parse beta-factor at line 10983 @> WARNING failed to parse beta-factor at line 10984 @> WARNING failed to parse beta-factor at line 10985 @> WARNING failed to parse beta-factor at line 10986 @> WARNING failed to parse beta-factor at line 10987 @> WARNING failed to parse beta-factor at line 10988 @> WARNING failed to parse beta-factor at line 10989 @> WARNING failed to parse beta-factor at line 10990 @> WARNING failed to parse beta-factor at line 10991 @> WARNING failed to parse beta-factor at line 10992 @> WARNING failed to parse beta-factor at line 10993 @> WARNING failed to parse beta-factor at line 10994 @> WARNING failed to parse beta-factor at line 10995 @> WARNING failed to parse beta-factor at line 10996 @> WARNING failed to parse beta-factor at line 10997 @> WARNING failed to parse beta-factor at line 10998 @> WARNING failed to parse beta-factor at line 10999 @> WARNING failed to parse beta-factor at line 11000 @> WARNING failed to parse beta-factor at line 11001 @> WARNING failed to parse beta-factor at line 11002 @> WARNING failed to parse beta-factor at line 11003 @> WARNING failed to parse beta-factor at line 11004 @> WARNING failed to parse beta-factor at line 11005 @> WARNING failed to parse beta-factor at line 11006 @> WARNING failed to parse beta-factor at line 11007 @> WARNING failed to parse beta-factor at line 11008 @> WARNING failed to parse beta-factor at line 11009 @> WARNING failed to parse beta-factor at line 11010 @> WARNING failed to parse beta-factor at line 11011 @> WARNING failed to parse beta-factor at line 11012 @> WARNING failed to parse beta-factor at line 11013 @> WARNING failed to parse beta-factor at line 11014 @> WARNING failed to parse beta-factor at line 11015 @> WARNING failed to parse beta-factor at line 11016 @> WARNING failed to parse beta-factor at line 11017 @> WARNING failed to parse beta-factor at line 11018 @> WARNING failed to parse beta-factor at line 11019 @> WARNING failed to parse beta-factor at line 11020 @> WARNING failed to parse beta-factor at line 11021 @> WARNING failed to parse beta-factor at line 11022 @> WARNING failed to parse beta-factor at line 11023 @> WARNING failed to parse beta-factor at line 11024 @> WARNING failed to parse beta-factor at line 11025 @> WARNING failed to parse beta-factor at line 11026 @> WARNING failed to parse beta-factor at line 11027 @> WARNING failed to parse beta-factor at line 11028 @> WARNING failed to parse beta-factor at line 11029 @> WARNING failed to parse beta-factor at line 11030 @> WARNING failed to parse beta-factor at line 11031 @> WARNING failed to parse beta-factor at line 11032 @> WARNING failed to parse beta-factor at line 11033 @> WARNING failed to parse beta-factor at line 11034 @> WARNING failed to parse beta-factor at line 11035 @> WARNING failed to parse beta-factor at line 11036 @> WARNING failed to parse beta-factor at line 11037 @> WARNING failed to parse beta-factor at line 11038 @> WARNING failed to parse beta-factor at line 11039 @> WARNING failed to parse beta-factor at line 11040 @> WARNING failed to parse beta-factor at line 11041 @> WARNING failed to parse beta-factor at line 11042 @> WARNING failed to parse beta-factor at line 11043 @> WARNING failed to parse beta-factor at line 11044 @> WARNING failed to parse beta-factor at line 11045 @> WARNING failed to parse beta-factor at line 11046 @> WARNING failed to parse beta-factor at line 11047 @> WARNING failed to parse beta-factor at line 11048 @> WARNING failed to parse beta-factor at line 11049 @> WARNING failed to parse beta-factor at line 11050 @> WARNING failed to parse beta-factor at line 11051 @> WARNING failed to parse beta-factor at line 11052 @> WARNING failed to parse beta-factor at line 11053 @> WARNING failed to parse beta-factor at line 11054 @> WARNING failed to parse beta-factor at line 11055 @> WARNING failed to parse beta-factor at line 11056 @> WARNING failed to parse beta-factor at line 11057 @> WARNING failed to parse beta-factor at line 11058 @> WARNING failed to parse beta-factor at line 11059 @> WARNING failed to parse beta-factor at line 11060 @> WARNING failed to parse beta-factor at line 11061 @> WARNING failed to parse beta-factor at line 11062 @> WARNING failed to parse beta-factor at line 11063 @> WARNING failed to parse beta-factor at line 11064 @> WARNING failed to parse beta-factor at line 11065 @> WARNING failed to parse beta-factor at line 11066 @> WARNING failed to parse beta-factor at line 11067 @> WARNING failed to parse beta-factor at line 11068 @> WARNING failed to parse beta-factor at line 11069 @> WARNING failed to parse beta-factor at line 11070 @> WARNING failed to parse beta-factor at line 11071 @> WARNING failed to parse beta-factor at line 11072 @> WARNING failed to parse beta-factor at line 11073 @> WARNING failed to parse beta-factor at line 11074 @> WARNING failed to parse beta-factor at line 11075 @> WARNING failed to parse beta-factor at line 11076 @> WARNING failed to parse beta-factor at line 11077 @> WARNING failed to parse beta-factor at line 11078 @> WARNING failed to parse beta-factor at line 11079 @> WARNING failed to parse beta-factor at line 11080 @> WARNING failed to parse beta-factor at line 11081 @> WARNING failed to parse beta-factor at line 11082 @> WARNING failed to parse beta-factor at line 11083 @> WARNING failed to parse beta-factor at line 11084 @> WARNING failed to parse beta-factor at line 11085 @> WARNING failed to parse beta-factor at line 11086 @> WARNING failed to parse beta-factor at line 11087 @> WARNING failed to parse beta-factor at line 11088 @> WARNING failed to parse beta-factor at line 11089 @> WARNING failed to parse beta-factor at line 11090 @> WARNING failed to parse beta-factor at line 11091 @> WARNING failed to parse beta-factor at line 11092 @> WARNING failed to parse beta-factor at line 11093 @> WARNING failed to parse beta-factor at line 11094 @> WARNING failed to parse beta-factor at line 11095 @> WARNING failed to parse beta-factor at line 11096 @> WARNING failed to parse beta-factor at line 11097 @> WARNING failed to parse beta-factor at line 11098 @> WARNING failed to parse beta-factor at line 11099 @> WARNING failed to parse beta-factor at line 11100 @> WARNING failed to parse beta-factor at line 11101 @> WARNING failed to parse beta-factor at line 11102 @> WARNING failed to parse beta-factor at line 11103 @> WARNING failed to parse beta-factor at line 11104 @> WARNING failed to parse beta-factor at line 11105 @> WARNING failed to parse beta-factor at line 11106 @> WARNING failed to parse beta-factor at line 11107 @> WARNING failed to parse beta-factor at line 11108 @> WARNING failed to parse beta-factor at line 11109 @> WARNING failed to parse beta-factor at line 11110 @> WARNING failed to parse beta-factor at line 11111 @> WARNING failed to parse beta-factor at line 11112 @> WARNING failed to parse beta-factor at line 11113 @> WARNING failed to parse beta-factor at line 11114 @> WARNING failed to parse beta-factor at line 11115 @> WARNING failed to parse beta-factor at line 11116 @> WARNING failed to parse beta-factor at line 11117 @> WARNING failed to parse beta-factor at line 11118 @> WARNING failed to parse beta-factor at line 11119 @> WARNING failed to parse beta-factor at line 11120 @> WARNING failed to parse beta-factor at line 11121 @> WARNING failed to parse beta-factor at line 11122 @> WARNING failed to parse beta-factor at line 11123 @> WARNING failed to parse beta-factor at line 11124 @> WARNING failed to parse beta-factor at line 11125 @> WARNING failed to parse beta-factor at line 11126 @> WARNING failed to parse beta-factor at line 11127 @> WARNING failed to parse beta-factor at line 11128 @> WARNING failed to parse beta-factor at line 11129 @> WARNING failed to parse beta-factor at line 11130 @> WARNING failed to parse beta-factor at line 11131 @> WARNING failed to parse beta-factor at line 11132 @> WARNING failed to parse beta-factor at line 11133 @> WARNING failed to parse beta-factor at line 11134 @> WARNING failed to parse beta-factor at line 11135 @> WARNING failed to parse beta-factor at line 11136 @> WARNING failed to parse beta-factor at line 11137 @> WARNING failed to parse beta-factor at line 11138 @> WARNING failed to parse beta-factor at line 11139 @> WARNING failed to parse beta-factor at line 11140 @> WARNING failed to parse beta-factor at line 11141 @> WARNING failed to parse beta-factor at line 11142 @> WARNING failed to parse beta-factor at line 11143 @> WARNING failed to parse beta-factor at line 11144 @> WARNING failed to parse beta-factor at line 11145 @> WARNING failed to parse beta-factor at line 11146 @> WARNING failed to parse beta-factor at line 11147 @> WARNING failed to parse beta-factor at line 11148 @> WARNING failed to parse beta-factor at line 11149 @> WARNING failed to parse beta-factor at line 11150 @> WARNING failed to parse beta-factor at line 11151 @> WARNING failed to parse beta-factor at line 11152 @> WARNING failed to parse beta-factor at line 11153 @> WARNING failed to parse beta-factor at line 11154 @> WARNING failed to parse beta-factor at line 11155 @> WARNING failed to parse beta-factor at line 11156 @> WARNING failed to parse beta-factor at line 11157 @> WARNING failed to parse beta-factor at line 11158 @> WARNING failed to parse beta-factor at line 11159 @> WARNING failed to parse beta-factor at line 11160 @> WARNING failed to parse beta-factor at line 11161 @> WARNING failed to parse beta-factor at line 11162 @> WARNING failed to parse beta-factor at line 11163 @> WARNING failed to parse beta-factor at line 11164 @> WARNING failed to parse beta-factor at line 11165 @> WARNING failed to parse beta-factor at line 11166 @> WARNING failed to parse beta-factor at line 11167 @> WARNING failed to parse beta-factor at line 11168 @> WARNING failed to parse beta-factor at line 11169 @> WARNING failed to parse beta-factor at line 11170 @> WARNING failed to parse beta-factor at line 11171 @> WARNING failed to parse beta-factor at line 11172 @> WARNING failed to parse beta-factor at line 11173 @> WARNING failed to parse beta-factor at line 11174 @> WARNING failed to parse beta-factor at line 11175 @> WARNING failed to parse beta-factor at line 11176 @> WARNING failed to parse beta-factor at line 11177 @> WARNING failed to parse beta-factor at line 11178 @> WARNING failed to parse beta-factor at line 11179 @> WARNING failed to parse beta-factor at line 11180 @> WARNING failed to parse beta-factor at line 11181 @> WARNING failed to parse beta-factor at line 11182 @> WARNING failed to parse beta-factor at line 11183 @> WARNING failed to parse beta-factor at line 11184 @> WARNING failed to parse beta-factor at line 11185 @> WARNING failed to parse beta-factor at line 11186 @> WARNING failed to parse beta-factor at line 11187 @> WARNING failed to parse beta-factor at line 11188 @> WARNING failed to parse beta-factor at line 11189 @> WARNING failed to parse beta-factor at line 11190 @> WARNING failed to parse beta-factor at line 11191 @> WARNING failed to parse beta-factor at line 11192 @> WARNING failed to parse beta-factor at line 11193 @> WARNING failed to parse beta-factor at line 11194 @> WARNING failed to parse beta-factor at line 11195 @> WARNING failed to parse beta-factor at line 11196 @> WARNING failed to parse beta-factor at line 11197 @> WARNING failed to parse beta-factor at line 11198 @> WARNING failed to parse beta-factor at line 11199 @> WARNING failed to parse beta-factor at line 11200 @> WARNING failed to parse beta-factor at line 11201 @> WARNING failed to parse beta-factor at line 11202 @> WARNING failed to parse beta-factor at line 11203 @> WARNING failed to parse beta-factor at line 11204 @> WARNING failed to parse beta-factor at line 11205 @> WARNING failed to parse beta-factor at line 11206 @> WARNING failed to parse beta-factor at line 11207 @> WARNING failed to parse beta-factor at line 11208 @> WARNING failed to parse beta-factor at line 11209 @> WARNING failed to parse beta-factor at line 11210 @> WARNING failed to parse beta-factor at line 11211 @> WARNING failed to parse beta-factor at line 11212 @> WARNING failed to parse beta-factor at line 11213 @> WARNING failed to parse beta-factor at line 11214 @> WARNING failed to parse beta-factor at line 11215 @> WARNING failed to parse beta-factor at line 11216 @> WARNING failed to parse beta-factor at line 11217 @> WARNING failed to parse beta-factor at line 11218 @> WARNING failed to parse beta-factor at line 11219 @> WARNING failed to parse beta-factor at line 11220 @> WARNING failed to parse beta-factor at line 11221 @> WARNING failed to parse beta-factor at line 11222 @> WARNING failed to parse beta-factor at line 11223 @> WARNING failed to parse beta-factor at line 11224 @> WARNING failed to parse beta-factor at line 11225 @> WARNING failed to parse beta-factor at line 11226 @> WARNING failed to parse beta-factor at line 11227 @> WARNING failed to parse beta-factor at line 11228 @> WARNING failed to parse beta-factor at line 11229 @> WARNING failed to parse beta-factor at line 11230 @> WARNING failed to parse beta-factor at line 11231 @> WARNING failed to parse beta-factor at line 11232 @> WARNING failed to parse beta-factor at line 11233 @> WARNING failed to parse beta-factor at line 11234 @> WARNING failed to parse beta-factor at line 11235 @> WARNING failed to parse beta-factor at line 11236 @> WARNING failed to parse beta-factor at line 11237 @> WARNING failed to parse beta-factor at line 11238 @> WARNING failed to parse beta-factor at line 11239 @> WARNING failed to parse beta-factor at line 11240 @> WARNING failed to parse beta-factor at line 11241 @> WARNING failed to parse beta-factor at line 11242 @> WARNING failed to parse beta-factor at line 11243 @> WARNING failed to parse beta-factor at line 11244 @> WARNING failed to parse beta-factor at line 11245 @> WARNING failed to parse beta-factor at line 11246 @> WARNING failed to parse beta-factor at line 11247 @> WARNING failed to parse beta-factor at line 11248 @> WARNING failed to parse beta-factor at line 11249 @> WARNING failed to parse beta-factor at line 11250 @> WARNING failed to parse beta-factor at line 11251 @> WARNING failed to parse beta-factor at line 11252 @> WARNING failed to parse beta-factor at line 11253 @> WARNING failed to parse beta-factor at line 11254 @> WARNING failed to parse beta-factor at line 11255 @> WARNING failed to parse beta-factor at line 11256 @> WARNING failed to parse beta-factor at line 11257 @> WARNING failed to parse beta-factor at line 11258 @> WARNING failed to parse beta-factor at line 11259 @> WARNING failed to parse beta-factor at line 11260 @> WARNING failed to parse beta-factor at line 11261 @> WARNING failed to parse beta-factor at line 11262 @> WARNING failed to parse beta-factor at line 11263 @> WARNING failed to parse beta-factor at line 11264 @> WARNING failed to parse beta-factor at line 11265 @> WARNING failed to parse beta-factor at line 11266 @> WARNING failed to parse beta-factor at line 11267 @> WARNING failed to parse beta-factor at line 11268 @> WARNING failed to parse beta-factor at line 11269 @> WARNING failed to parse beta-factor at line 11270 @> WARNING failed to parse beta-factor at line 11271 @> WARNING failed to parse beta-factor at line 11272 @> WARNING failed to parse beta-factor at line 11273 @> WARNING failed to parse beta-factor at line 11274 @> WARNING failed to parse beta-factor at line 11275 @> WARNING failed to parse beta-factor at line 11276 @> WARNING failed to parse beta-factor at line 11277 @> WARNING failed to parse beta-factor at line 11278 @> WARNING failed to parse beta-factor at line 11279 @> WARNING failed to parse beta-factor at line 11280 @> WARNING failed to parse beta-factor at line 11281 @> WARNING failed to parse beta-factor at line 11282 @> WARNING failed to parse beta-factor at line 11283 @> WARNING failed to parse beta-factor at line 11284 @> WARNING failed to parse beta-factor at line 11285 @> WARNING failed to parse beta-factor at line 11286 @> WARNING failed to parse beta-factor at line 11287 @> WARNING failed to parse beta-factor at line 11288 @> WARNING failed to parse beta-factor at line 11289 @> WARNING failed to parse beta-factor at line 11290 @> WARNING failed to parse beta-factor at line 11291 @> WARNING failed to parse beta-factor at line 11292 @> WARNING failed to parse beta-factor at line 11293 @> WARNING failed to parse beta-factor at line 11294 @> WARNING failed to parse beta-factor at line 11295 @> WARNING failed to parse beta-factor at line 11296 @> WARNING failed to parse beta-factor at line 11297 @> WARNING failed to parse beta-factor at line 11298 @> WARNING failed to parse beta-factor at line 11299 @> WARNING failed to parse beta-factor at line 11300 @> WARNING failed to parse beta-factor at line 11301 @> WARNING failed to parse beta-factor at line 11302 @> WARNING failed to parse beta-factor at line 11303 @> WARNING failed to parse beta-factor at line 11304 @> WARNING failed to parse beta-factor at line 11305 @> WARNING failed to parse beta-factor at line 11306 @> WARNING failed to parse beta-factor at line 11307 @> WARNING failed to parse beta-factor at line 11308 @> WARNING failed to parse beta-factor at line 11309 @> WARNING failed to parse beta-factor at line 11310 @> WARNING failed to parse beta-factor at line 11311 @> WARNING failed to parse beta-factor at line 11312 @> WARNING failed to parse beta-factor at line 11313 @> WARNING failed to parse beta-factor at line 11314 @> WARNING failed to parse beta-factor at line 11315 @> WARNING failed to parse beta-factor at line 11316 @> WARNING failed to parse beta-factor at line 11317 @> WARNING failed to parse beta-factor at line 11318 @> WARNING failed to parse beta-factor at line 11319 @> WARNING failed to parse beta-factor at line 11320 @> WARNING failed to parse beta-factor at line 11321 @> WARNING failed to parse beta-factor at line 11322 @> WARNING failed to parse beta-factor at line 11323 @> WARNING failed to parse beta-factor at line 11324 @> WARNING failed to parse beta-factor at line 11325 @> WARNING failed to parse beta-factor at line 11326 @> WARNING failed to parse beta-factor at line 11327 @> WARNING failed to parse beta-factor at line 11328 @> WARNING failed to parse beta-factor at line 11329 @> WARNING failed to parse beta-factor at line 11330 @> WARNING failed to parse beta-factor at line 11331 @> WARNING failed to parse beta-factor at line 11332 @> WARNING failed to parse beta-factor at line 11333 @> WARNING failed to parse beta-factor at line 11334 @> WARNING failed to parse beta-factor at line 11335 @> WARNING failed to parse beta-factor at line 11336 @> WARNING failed to parse beta-factor at line 11337 @> WARNING failed to parse beta-factor at line 11338 @> WARNING failed to parse beta-factor at line 11339 @> WARNING failed to parse beta-factor at line 11340 @> WARNING failed to parse beta-factor at line 11341 @> WARNING failed to parse beta-factor at line 11342 @> WARNING failed to parse beta-factor at line 11343 @> WARNING failed to parse beta-factor at line 11344 @> WARNING failed to parse beta-factor at line 11345 @> WARNING failed to parse beta-factor at line 11346 @> WARNING failed to parse beta-factor at line 11347 @> WARNING failed to parse beta-factor at line 11348 @> WARNING failed to parse beta-factor at line 11349 @> WARNING failed to parse beta-factor at line 11350 @> WARNING failed to parse beta-factor at line 11351 @> WARNING failed to parse beta-factor at line 11352 @> WARNING failed to parse beta-factor at line 11353 @> WARNING failed to parse beta-factor at line 11354 @> WARNING failed to parse beta-factor at line 11355 @> WARNING failed to parse beta-factor at line 11356 @> WARNING failed to parse beta-factor at line 11357 @> WARNING failed to parse beta-factor at line 11358 @> WARNING failed to parse beta-factor at line 11359 @> WARNING failed to parse beta-factor at line 11360 @> WARNING failed to parse beta-factor at line 11361 @> WARNING failed to parse beta-factor at line 11362 @> WARNING failed to parse beta-factor at line 11363 @> WARNING failed to parse beta-factor at line 11364 @> WARNING failed to parse beta-factor at line 11365 @> WARNING failed to parse beta-factor at line 11366 @> WARNING failed to parse beta-factor at line 11367 @> WARNING failed to parse beta-factor at line 11368 @> WARNING failed to parse beta-factor at line 11369 @> WARNING failed to parse beta-factor at line 11370 @> WARNING failed to parse beta-factor at line 11371 @> WARNING failed to parse beta-factor at line 11372 @> WARNING failed to parse beta-factor at line 11373 @> WARNING failed to parse beta-factor at line 11374 @> WARNING failed to parse beta-factor at line 11375 @> WARNING failed to parse beta-factor at line 11376 @> WARNING failed to parse beta-factor at line 11377 @> WARNING failed to parse beta-factor at line 11378 @> WARNING failed to parse beta-factor at line 11379 @> WARNING failed to parse beta-factor at line 11380 @> WARNING failed to parse beta-factor at line 11381 @> WARNING failed to parse beta-factor at line 11382 @> WARNING failed to parse beta-factor at line 11383 @> WARNING failed to parse beta-factor at line 11384 @> WARNING failed to parse beta-factor at line 11385 @> WARNING failed to parse beta-factor at line 11386 @> WARNING failed to parse beta-factor at line 11387 @> WARNING failed to parse beta-factor at line 11388 @> WARNING failed to parse beta-factor at line 11389 @> WARNING failed to parse beta-factor at line 11390 @> WARNING failed to parse beta-factor at line 11391 @> WARNING failed to parse beta-factor at line 11392 @> WARNING failed to parse beta-factor at line 11393 @> WARNING failed to parse beta-factor at line 11394 @> WARNING failed to parse beta-factor at line 11395 @> WARNING failed to parse beta-factor at line 11396 @> WARNING failed to parse beta-factor at line 11397 @> WARNING failed to parse beta-factor at line 11398 @> WARNING failed to parse beta-factor at line 11399 @> WARNING failed to parse beta-factor at line 11400 @> WARNING failed to parse beta-factor at line 11401 @> WARNING failed to parse beta-factor at line 11402 @> WARNING failed to parse beta-factor at line 11403 @> WARNING failed to parse beta-factor at line 11404 @> WARNING failed to parse beta-factor at line 11405 @> WARNING failed to parse beta-factor at line 11406 @> WARNING failed to parse beta-factor at line 11407 @> WARNING failed to parse beta-factor at line 11408 @> WARNING failed to parse beta-factor at line 11409 @> WARNING failed to parse beta-factor at line 11410 @> WARNING failed to parse beta-factor at line 11411 @> WARNING failed to parse beta-factor at line 11412 @> WARNING failed to parse beta-factor at line 11413 @> WARNING failed to parse beta-factor at line 11414 @> WARNING failed to parse beta-factor at line 11415 @> WARNING failed to parse beta-factor at line 11416 @> WARNING failed to parse beta-factor at line 11417 @> WARNING failed to parse beta-factor at line 11418 @> WARNING failed to parse beta-factor at line 11419 @> WARNING failed to parse beta-factor at line 11420 @> WARNING failed to parse beta-factor at line 11421 @> WARNING failed to parse beta-factor at line 11422 @> WARNING failed to parse beta-factor at line 11423 @> WARNING failed to parse beta-factor at line 11424 @> WARNING failed to parse beta-factor at line 11425 @> WARNING failed to parse beta-factor at line 11426 @> WARNING failed to parse beta-factor at line 11427 @> WARNING failed to parse beta-factor at line 11428 @> WARNING failed to parse beta-factor at line 11429 @> WARNING failed to parse beta-factor at line 11430 @> WARNING failed to parse beta-factor at line 11431 @> WARNING failed to parse beta-factor at line 11432 @> WARNING failed to parse beta-factor at line 11433 @> WARNING failed to parse beta-factor at line 11434 @> WARNING failed to parse beta-factor at line 11435 @> WARNING failed to parse beta-factor at line 11436 @> WARNING failed to parse beta-factor at line 11437 @> WARNING failed to parse beta-factor at line 11438 @> WARNING failed to parse beta-factor at line 11439 @> WARNING failed to parse beta-factor at line 11440 @> WARNING failed to parse beta-factor at line 11441 @> WARNING failed to parse beta-factor at line 11442 @> WARNING failed to parse beta-factor at line 11443 @> WARNING failed to parse beta-factor at line 11444 @> WARNING failed to parse beta-factor at line 11445 @> WARNING failed to parse beta-factor at line 11446 @> WARNING failed to parse beta-factor at line 11447 @> WARNING failed to parse beta-factor at line 11448 @> WARNING failed to parse beta-factor at line 11449 @> WARNING failed to parse beta-factor at line 11450 @> WARNING failed to parse beta-factor at line 11451 @> WARNING failed to parse beta-factor at line 11452 @> WARNING failed to parse beta-factor at line 11453 @> WARNING failed to parse beta-factor at line 11454 @> WARNING failed to parse beta-factor at line 11455 @> WARNING failed to parse beta-factor at line 11456 @> WARNING failed to parse beta-factor at line 11457 @> WARNING failed to parse beta-factor at line 11458 @> WARNING failed to parse beta-factor at line 11459 @> WARNING failed to parse beta-factor at line 11460 @> WARNING failed to parse beta-factor at line 11461 @> WARNING failed to parse beta-factor at line 11462 @> WARNING failed to parse beta-factor at line 11463 @> WARNING failed to parse beta-factor at line 11464 @> WARNING failed to parse beta-factor at line 11465 @> WARNING failed to parse beta-factor at line 11466 @> WARNING failed to parse beta-factor at line 11467 @> WARNING failed to parse beta-factor at line 11468 @> WARNING failed to parse beta-factor at line 11469 @> WARNING failed to parse beta-factor at line 11470 @> WARNING failed to parse beta-factor at line 11471 @> WARNING failed to parse beta-factor at line 11472 @> WARNING failed to parse beta-factor at line 11473 @> WARNING failed to parse beta-factor at line 11474 @> WARNING failed to parse beta-factor at line 11475 @> WARNING failed to parse beta-factor at line 11476 @> WARNING failed to parse beta-factor at line 11477 @> WARNING failed to parse beta-factor at line 11478 @> WARNING failed to parse beta-factor at line 11479 @> WARNING failed to parse beta-factor at line 11480 @> WARNING failed to parse beta-factor at line 11481 @> WARNING failed to parse beta-factor at line 11482 @> WARNING failed to parse beta-factor at line 11483 @> WARNING failed to parse beta-factor at line 11484 @> WARNING failed to parse beta-factor at line 11485 @> WARNING failed to parse beta-factor at line 11486 @> WARNING failed to parse beta-factor at line 11487 @> WARNING failed to parse beta-factor at line 11488 @> WARNING failed to parse beta-factor at line 11489 @> WARNING failed to parse beta-factor at line 11490 @> WARNING failed to parse beta-factor at line 11491 @> WARNING failed to parse beta-factor at line 11492 @> WARNING failed to parse beta-factor at line 11493 @> WARNING failed to parse beta-factor at line 11494 @> WARNING failed to parse beta-factor at line 11495 @> WARNING failed to parse beta-factor at line 11496 @> WARNING failed to parse beta-factor at line 11497 @> WARNING failed to parse beta-factor at line 11498 @> WARNING failed to parse beta-factor at line 11499 @> WARNING failed to parse beta-factor at line 11500 @> WARNING failed to parse beta-factor at line 11501 @> WARNING failed to parse beta-factor at line 11502 @> WARNING failed to parse beta-factor at line 11503 @> WARNING failed to parse beta-factor at line 11504 @> WARNING failed to parse beta-factor at line 11505 @> WARNING failed to parse beta-factor at line 11506 @> WARNING failed to parse beta-factor at line 11507 @> WARNING failed to parse beta-factor at line 11508 @> WARNING failed to parse beta-factor at line 11509 @> WARNING failed to parse beta-factor at line 11510 @> WARNING failed to parse beta-factor at line 11511 @> WARNING failed to parse beta-factor at line 11512 @> WARNING failed to parse beta-factor at line 11513 @> WARNING failed to parse beta-factor at line 11514 @> WARNING failed to parse beta-factor at line 11515 @> WARNING failed to parse beta-factor at line 11516 @> WARNING failed to parse beta-factor at line 11517 @> WARNING failed to parse beta-factor at line 11518 @> WARNING failed to parse beta-factor at line 11519 @> WARNING failed to parse beta-factor at line 11520 @> WARNING failed to parse beta-factor at line 11521 @> WARNING failed to parse beta-factor at line 11522 @> WARNING failed to parse beta-factor at line 11523 @> WARNING failed to parse beta-factor at line 11524 @> WARNING failed to parse beta-factor at line 11525 @> WARNING failed to parse beta-factor at line 11526 @> WARNING failed to parse beta-factor at line 11527 @> WARNING failed to parse beta-factor at line 11528 @> WARNING failed to parse beta-factor at line 11529 @> WARNING failed to parse beta-factor at line 11530 @> WARNING failed to parse beta-factor at line 11531 @> WARNING failed to parse beta-factor at line 11532 @> WARNING failed to parse beta-factor at line 11533 @> WARNING failed to parse beta-factor at line 11534 @> WARNING failed to parse beta-factor at line 11535 @> WARNING failed to parse beta-factor at line 11536 @> WARNING failed to parse beta-factor at line 11537 @> WARNING failed to parse beta-factor at line 11538 @> WARNING failed to parse beta-factor at line 11539 @> WARNING failed to parse beta-factor at line 11540 @> WARNING failed to parse beta-factor at line 11541 @> WARNING failed to parse beta-factor at line 11542 @> WARNING failed to parse beta-factor at line 11543 @> WARNING failed to parse beta-factor at line 11544 @> WARNING failed to parse beta-factor at line 11545 @> WARNING failed to parse beta-factor at line 11546 @> WARNING failed to parse beta-factor at line 11547 @> WARNING failed to parse beta-factor at line 11548 @> WARNING failed to parse beta-factor at line 11549 @> WARNING failed to parse beta-factor at line 11550 @> WARNING failed to parse beta-factor at line 11551 @> WARNING failed to parse beta-factor at line 11552 @> WARNING failed to parse beta-factor at line 11553 @> WARNING failed to parse beta-factor at line 11554 @> WARNING failed to parse beta-factor at line 11555 @> WARNING failed to parse beta-factor at line 11556 @> WARNING failed to parse beta-factor at line 11557 @> WARNING failed to parse beta-factor at line 11558 @> WARNING failed to parse beta-factor at line 11559 @> WARNING failed to parse beta-factor at line 11560 @> WARNING failed to parse beta-factor at line 11561 @> WARNING failed to parse beta-factor at line 11562 @> WARNING failed to parse beta-factor at line 11563 @> WARNING failed to parse beta-factor at line 11564 @> WARNING failed to parse beta-factor at line 11565 @> WARNING failed to parse beta-factor at line 11566 @> WARNING failed to parse beta-factor at line 11567 @> WARNING failed to parse beta-factor at line 11568 @> WARNING failed to parse beta-factor at line 11569 @> WARNING failed to parse beta-factor at line 11570 @> WARNING failed to parse beta-factor at line 11571 @> WARNING failed to parse beta-factor at line 11572 @> WARNING failed to parse beta-factor at line 11573 @> WARNING failed to parse beta-factor at line 11574 @> WARNING failed to parse beta-factor at line 11575 @> WARNING failed to parse beta-factor at line 11576 @> WARNING failed to parse beta-factor at line 11577 @> WARNING failed to parse beta-factor at line 11578 @> WARNING failed to parse beta-factor at line 11579 @> WARNING failed to parse beta-factor at line 11580 @> WARNING failed to parse beta-factor at line 11581 @> WARNING failed to parse beta-factor at line 11582 @> WARNING failed to parse beta-factor at line 11583 @> WARNING failed to parse beta-factor at line 11584 @> WARNING failed to parse beta-factor at line 11585 @> WARNING failed to parse beta-factor at line 11586 @> WARNING failed to parse beta-factor at line 11587 @> WARNING failed to parse beta-factor at line 11588 @> WARNING failed to parse beta-factor at line 11589 @> WARNING failed to parse beta-factor at line 11590 @> WARNING failed to parse beta-factor at line 11591 @> WARNING failed to parse beta-factor at line 11592 @> WARNING failed to parse beta-factor at line 11593 @> WARNING failed to parse beta-factor at line 11594 @> WARNING failed to parse beta-factor at line 11595 @> WARNING failed to parse beta-factor at line 11596 @> WARNING failed to parse beta-factor at line 11597 @> WARNING failed to parse beta-factor at line 11598 @> WARNING failed to parse beta-factor at line 11599 @> WARNING failed to parse beta-factor at line 11600 @> WARNING failed to parse beta-factor at line 11601 @> WARNING failed to parse beta-factor at line 11602 @> WARNING failed to parse beta-factor at line 11603 @> WARNING failed to parse beta-factor at line 11604 @> WARNING failed to parse beta-factor at line 11605 @> WARNING failed to parse beta-factor at line 11606 @> WARNING failed to parse beta-factor at line 11607 @> WARNING failed to parse beta-factor at line 11608 @> WARNING failed to parse beta-factor at line 11609 @> WARNING failed to parse beta-factor at line 11610 @> WARNING failed to parse beta-factor at line 11611 @> WARNING failed to parse beta-factor at line 11612 @> WARNING failed to parse beta-factor at line 11613 @> WARNING failed to parse beta-factor at line 11614 @> WARNING failed to parse beta-factor at line 11615 @> WARNING failed to parse beta-factor at line 11616 @> WARNING failed to parse beta-factor at line 11617 @> WARNING failed to parse beta-factor at line 11618 @> WARNING failed to parse beta-factor at line 11619 @> WARNING failed to parse beta-factor at line 11620 @> WARNING failed to parse beta-factor at line 11621 @> WARNING failed to parse beta-factor at line 11622 @> WARNING failed to parse beta-factor at line 11623 @> WARNING failed to parse beta-factor at line 11624 @> WARNING failed to parse beta-factor at line 11625 @> WARNING failed to parse beta-factor at line 11626 @> WARNING failed to parse beta-factor at line 11627 @> WARNING failed to parse beta-factor at line 11628 @> WARNING failed to parse beta-factor at line 11629 @> WARNING failed to parse beta-factor at line 11630 @> WARNING failed to parse beta-factor at line 11631 @> WARNING failed to parse beta-factor at line 11632 @> WARNING failed to parse beta-factor at line 11633 @> WARNING failed to parse beta-factor at line 11634 @> WARNING failed to parse beta-factor at line 11635 @> WARNING failed to parse beta-factor at line 11636 @> WARNING failed to parse beta-factor at line 11637 @> WARNING failed to parse beta-factor at line 11638 @> WARNING failed to parse beta-factor at line 11639 @> WARNING failed to parse beta-factor at line 11640 @> WARNING failed to parse beta-factor at line 11641 @> WARNING failed to parse beta-factor at line 11642 @> WARNING failed to parse beta-factor at line 11643 @> WARNING failed to parse beta-factor at line 11644 @> WARNING failed to parse beta-factor at line 11645 @> WARNING failed to parse beta-factor at line 11646 @> WARNING failed to parse beta-factor at line 11647 @> WARNING failed to parse beta-factor at line 11648 @> WARNING failed to parse beta-factor at line 11649 @> WARNING failed to parse beta-factor at line 11650 @> WARNING failed to parse beta-factor at line 11651 @> WARNING failed to parse beta-factor at line 11652 @> WARNING failed to parse beta-factor at line 11653 @> WARNING failed to parse beta-factor at line 11654 @> WARNING failed to parse beta-factor at line 11655 @> WARNING failed to parse beta-factor at line 11656 @> WARNING failed to parse beta-factor at line 11657 @> WARNING failed to parse beta-factor at line 11658 @> WARNING failed to parse beta-factor at line 11659 @> WARNING failed to parse beta-factor at line 11660 @> WARNING failed to parse beta-factor at line 11661 @> WARNING failed to parse beta-factor at line 11662 @> WARNING failed to parse beta-factor at line 11663 @> WARNING failed to parse beta-factor at line 11664 @> WARNING failed to parse beta-factor at line 11665 @> WARNING failed to parse beta-factor at line 11666 @> WARNING failed to parse beta-factor at line 11667 @> WARNING failed to parse beta-factor at line 11668 @> WARNING failed to parse beta-factor at line 11669 @> WARNING failed to parse beta-factor at line 11670 @> WARNING failed to parse beta-factor at line 11671 @> WARNING failed to parse beta-factor at line 11672 @> WARNING failed to parse beta-factor at line 11673 @> WARNING failed to parse beta-factor at line 11674 @> WARNING failed to parse beta-factor at line 11675 @> WARNING failed to parse beta-factor at line 11676 @> WARNING failed to parse beta-factor at line 11677 @> WARNING failed to parse beta-factor at line 11678 @> WARNING failed to parse beta-factor at line 11679 @> WARNING failed to parse beta-factor at line 11680 @> WARNING failed to parse beta-factor at line 11681 @> WARNING failed to parse beta-factor at line 11682 @> WARNING failed to parse beta-factor at line 11683 @> WARNING failed to parse beta-factor at line 11684 @> WARNING failed to parse beta-factor at line 11685 @> WARNING failed to parse beta-factor at line 11686 @> WARNING failed to parse beta-factor at line 11687 @> WARNING failed to parse beta-factor at line 11688 @> WARNING failed to parse beta-factor at line 11689 @> WARNING failed to parse beta-factor at line 11690 @> WARNING failed to parse beta-factor at line 11691 @> WARNING failed to parse beta-factor at line 11692 @> WARNING failed to parse beta-factor at line 11693 @> WARNING failed to parse beta-factor at line 11694 @> WARNING failed to parse beta-factor at line 11695 @> WARNING failed to parse beta-factor at line 11696 @> WARNING failed to parse beta-factor at line 11697 @> WARNING failed to parse beta-factor at line 11698 @> WARNING failed to parse beta-factor at line 11699 @> WARNING failed to parse beta-factor at line 11700 @> WARNING failed to parse beta-factor at line 11701 @> WARNING failed to parse beta-factor at line 11702 @> WARNING failed to parse beta-factor at line 11703 @> WARNING failed to parse beta-factor at line 11704 @> WARNING failed to parse beta-factor at line 11705 @> WARNING failed to parse beta-factor at line 11706 @> WARNING failed to parse beta-factor at line 11707 @> WARNING failed to parse beta-factor at line 11708 @> WARNING failed to parse beta-factor at line 11709 @> WARNING failed to parse beta-factor at line 11710 @> WARNING failed to parse beta-factor at line 11711 @> WARNING failed to parse beta-factor at line 11712 @> WARNING failed to parse beta-factor at line 11713 @> WARNING failed to parse beta-factor at line 11714 @> WARNING failed to parse beta-factor at line 11715 @> WARNING failed to parse beta-factor at line 11716 @> WARNING failed to parse beta-factor at line 11717 @> WARNING failed to parse beta-factor at line 11718 @> WARNING failed to parse beta-factor at line 11719 @> WARNING failed to parse beta-factor at line 11720 @> WARNING failed to parse beta-factor at line 11721 @> WARNING failed to parse beta-factor at line 11722 @> WARNING failed to parse beta-factor at line 11723 @> WARNING failed to parse beta-factor at line 11724 @> WARNING failed to parse beta-factor at line 11725 @> WARNING failed to parse beta-factor at line 11726 @> WARNING failed to parse beta-factor at line 11727 @> WARNING failed to parse beta-factor at line 11728 @> WARNING failed to parse beta-factor at line 11729 @> WARNING failed to parse beta-factor at line 11730 @> WARNING failed to parse beta-factor at line 11731 @> WARNING failed to parse beta-factor at line 11732 @> WARNING failed to parse beta-factor at line 11733 @> WARNING failed to parse beta-factor at line 11734 @> WARNING failed to parse beta-factor at line 11735 @> WARNING failed to parse beta-factor at line 11736 @> WARNING failed to parse beta-factor at line 11737 @> WARNING failed to parse beta-factor at line 11738 @> WARNING failed to parse beta-factor at line 11739 @> WARNING failed to parse beta-factor at line 11740 @> WARNING failed to parse beta-factor at line 11741 @> WARNING failed to parse beta-factor at line 11742 @> WARNING failed to parse beta-factor at line 11743 @> WARNING failed to parse beta-factor at line 11744 @> WARNING failed to parse beta-factor at line 11745 @> WARNING failed to parse beta-factor at line 11746 @> WARNING failed to parse beta-factor at line 11747 @> WARNING failed to parse beta-factor at line 11748 @> WARNING failed to parse beta-factor at line 11749 @> WARNING failed to parse beta-factor at line 11750 @> WARNING failed to parse beta-factor at line 11751 @> WARNING failed to parse beta-factor at line 11752 @> WARNING failed to parse beta-factor at line 11753 @> WARNING failed to parse beta-factor at line 11754 @> WARNING failed to parse beta-factor at line 11755 @> WARNING failed to parse beta-factor at line 11756 @> WARNING failed to parse beta-factor at line 11757 @> WARNING failed to parse beta-factor at line 11758 @> WARNING failed to parse beta-factor at line 11759 @> WARNING failed to parse beta-factor at line 11760 @> WARNING failed to parse beta-factor at line 11761 @> WARNING failed to parse beta-factor at line 11762 @> WARNING failed to parse beta-factor at line 11763 @> WARNING failed to parse beta-factor at line 11764 @> WARNING failed to parse beta-factor at line 11765 @> WARNING failed to parse beta-factor at line 11766 @> WARNING failed to parse beta-factor at line 11767 @> WARNING failed to parse beta-factor at line 11768 @> WARNING failed to parse beta-factor at line 11769 @> WARNING failed to parse beta-factor at line 11770 @> WARNING failed to parse beta-factor at line 11771 @> WARNING failed to parse beta-factor at line 11772 @> WARNING failed to parse beta-factor at line 11773 @> WARNING failed to parse beta-factor at line 11774 @> WARNING failed to parse beta-factor at line 11775 @> WARNING failed to parse beta-factor at line 11776 @> WARNING failed to parse beta-factor at line 11777 @> WARNING failed to parse beta-factor at line 11778 @> WARNING failed to parse beta-factor at line 11779 @> WARNING failed to parse beta-factor at line 11780 @> WARNING failed to parse beta-factor at line 11781 @> WARNING failed to parse beta-factor at line 11782 @> WARNING failed to parse beta-factor at line 11783 @> WARNING failed to parse beta-factor at line 11784 @> WARNING failed to parse beta-factor at line 11785 @> WARNING failed to parse beta-factor at line 11786 @> WARNING failed to parse beta-factor at line 11787 @> WARNING failed to parse beta-factor at line 11788 @> WARNING failed to parse beta-factor at line 11789 @> WARNING failed to parse beta-factor at line 11790 @> WARNING failed to parse beta-factor at line 11791 @> WARNING failed to parse beta-factor at line 11792 @> WARNING failed to parse beta-factor at line 11793 @> WARNING failed to parse beta-factor at line 11794 @> WARNING failed to parse beta-factor at line 11795 @> WARNING failed to parse beta-factor at line 11796 @> WARNING failed to parse beta-factor at line 11797 @> WARNING failed to parse beta-factor at line 11798 @> WARNING failed to parse beta-factor at line 11799 @> WARNING failed to parse beta-factor at line 11800 @> WARNING failed to parse beta-factor at line 11801 @> WARNING failed to parse beta-factor at line 11802 @> WARNING failed to parse beta-factor at line 11803 @> WARNING failed to parse beta-factor at line 11804 @> WARNING failed to parse beta-factor at line 11805 @> WARNING failed to parse beta-factor at line 11806 @> WARNING failed to parse beta-factor at line 11807 @> WARNING failed to parse beta-factor at line 11808 @> WARNING failed to parse beta-factor at line 11809 @> WARNING failed to parse beta-factor at line 11810 @> WARNING failed to parse beta-factor at line 11811 @> WARNING failed to parse beta-factor at line 11812 @> WARNING failed to parse beta-factor at line 11813 @> WARNING failed to parse beta-factor at line 11814 @> WARNING failed to parse beta-factor at line 11815 @> WARNING failed to parse beta-factor at line 11816 @> WARNING failed to parse beta-factor at line 11817 @> WARNING failed to parse beta-factor at line 11818 @> WARNING failed to parse beta-factor at line 11819 @> WARNING failed to parse beta-factor at line 11820 @> WARNING failed to parse beta-factor at line 11821 @> WARNING failed to parse beta-factor at line 11822 @> WARNING failed to parse beta-factor at line 11823 @> WARNING failed to parse beta-factor at line 11824 @> WARNING failed to parse beta-factor at line 11825 @> WARNING failed to parse beta-factor at line 11826 @> WARNING failed to parse beta-factor at line 11827 @> WARNING failed to parse beta-factor at line 11828 @> WARNING failed to parse beta-factor at line 11829 @> WARNING failed to parse beta-factor at line 11830 @> WARNING failed to parse beta-factor at line 11831 @> WARNING failed to parse beta-factor at line 11832 @> WARNING failed to parse beta-factor at line 11833 @> WARNING failed to parse beta-factor at line 11834 @> WARNING failed to parse beta-factor at line 11835 @> WARNING failed to parse beta-factor at line 11836 @> WARNING failed to parse beta-factor at line 11837 @> WARNING failed to parse beta-factor at line 11838 @> WARNING failed to parse beta-factor at line 11839 @> WARNING failed to parse beta-factor at line 11840 @> WARNING failed to parse beta-factor at line 11841 @> WARNING failed to parse beta-factor at line 11842 @> WARNING failed to parse beta-factor at line 11843 @> WARNING failed to parse beta-factor at line 11844 @> WARNING failed to parse beta-factor at line 11845 @> WARNING failed to parse beta-factor at line 11846 @> WARNING failed to parse beta-factor at line 11847 @> WARNING failed to parse beta-factor at line 11848 @> WARNING failed to parse beta-factor at line 11849 @> WARNING failed to parse beta-factor at line 11850 @> WARNING failed to parse beta-factor at line 11851 @> WARNING failed to parse beta-factor at line 11852 @> WARNING failed to parse beta-factor at line 11853 @> WARNING failed to parse beta-factor at line 11854 @> WARNING failed to parse beta-factor at line 11855 @> WARNING failed to parse beta-factor at line 11856 @> WARNING failed to parse beta-factor at line 11857 @> WARNING failed to parse beta-factor at line 11858 @> WARNING failed to parse beta-factor at line 11859 @> WARNING failed to parse beta-factor at line 11860 @> WARNING failed to parse beta-factor at line 11861 @> WARNING failed to parse beta-factor at line 11862 @> WARNING failed to parse beta-factor at line 11863 @> WARNING failed to parse beta-factor at line 11864 @> WARNING failed to parse beta-factor at line 11865 @> WARNING failed to parse beta-factor at line 11866 @> WARNING failed to parse beta-factor at line 11867 @> WARNING failed to parse beta-factor at line 11868 @> WARNING failed to parse beta-factor at line 11869 @> WARNING failed to parse beta-factor at line 11870 @> WARNING failed to parse beta-factor at line 11871 @> WARNING failed to parse beta-factor at line 11872 @> WARNING failed to parse beta-factor at line 11873 @> WARNING failed to parse beta-factor at line 11874 @> WARNING failed to parse beta-factor at line 11875 @> WARNING failed to parse beta-factor at line 11876 @> WARNING failed to parse beta-factor at line 11877 @> WARNING failed to parse beta-factor at line 11878 @> WARNING failed to parse beta-factor at line 11879 @> WARNING failed to parse beta-factor at line 11880 @> WARNING failed to parse beta-factor at line 11881 @> WARNING failed to parse beta-factor at line 11882 @> WARNING failed to parse beta-factor at line 11883 @> WARNING failed to parse beta-factor at line 11884 @> WARNING failed to parse beta-factor at line 11885 @> WARNING failed to parse beta-factor at line 11886 @> WARNING failed to parse beta-factor at line 11887 @> WARNING failed to parse beta-factor at line 11888 @> WARNING failed to parse beta-factor at line 11889 @> WARNING failed to parse beta-factor at line 11890 @> WARNING failed to parse beta-factor at line 11891 @> WARNING failed to parse beta-factor at line 11892 @> WARNING failed to parse beta-factor at line 11893 @> WARNING failed to parse beta-factor at line 11894 @> WARNING failed to parse beta-factor at line 11895 @> WARNING failed to parse beta-factor at line 11896 @> WARNING failed to parse beta-factor at line 11897 @> WARNING failed to parse beta-factor at line 11898 @> WARNING failed to parse beta-factor at line 11899 @> WARNING failed to parse beta-factor at line 11900 @> WARNING failed to parse beta-factor at line 11901 @> WARNING failed to parse beta-factor at line 11902 @> WARNING failed to parse beta-factor at line 11903 @> WARNING failed to parse beta-factor at line 11904 @> WARNING failed to parse beta-factor at line 11905 @> WARNING failed to parse beta-factor at line 11906 @> WARNING failed to parse beta-factor at line 11907 @> WARNING failed to parse beta-factor at line 11908 @> WARNING failed to parse beta-factor at line 11909 @> WARNING failed to parse beta-factor at line 11910 @> WARNING failed to parse beta-factor at line 11911 @> WARNING failed to parse beta-factor at line 11912 @> WARNING failed to parse beta-factor at line 11913 @> WARNING failed to parse beta-factor at line 11914 @> WARNING failed to parse beta-factor at line 11915 @> WARNING failed to parse beta-factor at line 11916 @> WARNING failed to parse beta-factor at line 11917 @> WARNING failed to parse beta-factor at line 11918 @> WARNING failed to parse beta-factor at line 11919 @> WARNING failed to parse beta-factor at line 11920 @> WARNING failed to parse beta-factor at line 11921 @> WARNING failed to parse beta-factor at line 11922 @> WARNING failed to parse beta-factor at line 11923 @> WARNING failed to parse beta-factor at line 11924 @> WARNING failed to parse beta-factor at line 11925 @> WARNING failed to parse beta-factor at line 11926 @> WARNING failed to parse beta-factor at line 11927 @> WARNING failed to parse beta-factor at line 11928 @> WARNING failed to parse beta-factor at line 11929 @> WARNING failed to parse beta-factor at line 11930 @> WARNING failed to parse beta-factor at line 11931 @> WARNING failed to parse beta-factor at line 11932 @> WARNING failed to parse beta-factor at line 11933 @> WARNING failed to parse beta-factor at line 11934 @> WARNING failed to parse beta-factor at line 11935 @> WARNING failed to parse beta-factor at line 11936 @> WARNING failed to parse beta-factor at line 11937 @> WARNING failed to parse beta-factor at line 11938 @> WARNING failed to parse beta-factor at line 11939 @> WARNING failed to parse beta-factor at line 11940 @> WARNING failed to parse beta-factor at line 11941 @> WARNING failed to parse beta-factor at line 11942 @> WARNING failed to parse beta-factor at line 11943 @> WARNING failed to parse beta-factor at line 11944 @> WARNING failed to parse beta-factor at line 11945 @> WARNING failed to parse beta-factor at line 11946 @> WARNING failed to parse beta-factor at line 11947 @> WARNING failed to parse beta-factor at line 11948 @> WARNING failed to parse beta-factor at line 11949 @> WARNING failed to parse beta-factor at line 11950 @> WARNING failed to parse beta-factor at line 11951 @> WARNING failed to parse beta-factor at line 11952 @> WARNING failed to parse beta-factor at line 11953 @> WARNING failed to parse beta-factor at line 11954 @> WARNING failed to parse beta-factor at line 11955 @> WARNING failed to parse beta-factor at line 11956 @> WARNING failed to parse beta-factor at line 11957 @> WARNING failed to parse beta-factor at line 11958 @> WARNING failed to parse beta-factor at line 11959 @> WARNING failed to parse beta-factor at line 11960 @> WARNING failed to parse beta-factor at line 11961 @> WARNING failed to parse beta-factor at line 11962 @> WARNING failed to parse beta-factor at line 11963 @> WARNING failed to parse beta-factor at line 11964 @> WARNING failed to parse beta-factor at line 11965 @> WARNING failed to parse beta-factor at line 11966 @> WARNING failed to parse beta-factor at line 11967 @> WARNING failed to parse beta-factor at line 11968 @> WARNING failed to parse beta-factor at line 11969 @> WARNING failed to parse beta-factor at line 11970 @> WARNING failed to parse beta-factor at line 11971 @> WARNING failed to parse beta-factor at line 11972 @> WARNING failed to parse beta-factor at line 11973 @> WARNING failed to parse beta-factor at line 11974 @> WARNING failed to parse beta-factor at line 11975 @> WARNING failed to parse beta-factor at line 11976 @> WARNING failed to parse beta-factor at line 11977 @> WARNING failed to parse beta-factor at line 11978 @> WARNING failed to parse beta-factor at line 11979 @> WARNING failed to parse beta-factor at line 11980 @> WARNING failed to parse beta-factor at line 11981 @> WARNING failed to parse beta-factor at line 11982 @> WARNING failed to parse beta-factor at line 11983 @> WARNING failed to parse beta-factor at line 11984 @> WARNING failed to parse beta-factor at line 11985 @> WARNING failed to parse beta-factor at line 11986 @> WARNING failed to parse beta-factor at line 11987 @> WARNING failed to parse beta-factor at line 11988 @> WARNING failed to parse beta-factor at line 11989 @> WARNING failed to parse beta-factor at line 11990 @> WARNING failed to parse beta-factor at line 11991 @> WARNING failed to parse beta-factor at line 11992 @> WARNING failed to parse beta-factor at line 11993 @> WARNING failed to parse beta-factor at line 11994 @> WARNING failed to parse beta-factor at line 11995 @> WARNING failed to parse beta-factor at line 11996 @> WARNING failed to parse beta-factor at line 11997 @> WARNING failed to parse beta-factor at line 11998 @> WARNING failed to parse beta-factor at line 11999 @> WARNING failed to parse beta-factor at line 12000 @> WARNING failed to parse beta-factor at line 12001 @> WARNING failed to parse beta-factor at line 12002 @> WARNING failed to parse beta-factor at line 12003 @> WARNING failed to parse beta-factor at line 12004 @> WARNING failed to parse beta-factor at line 12005 @> WARNING failed to parse beta-factor at line 12006 @> WARNING failed to parse beta-factor at line 12007 @> WARNING failed to parse beta-factor at line 12008 @> WARNING failed to parse beta-factor at line 12009 @> WARNING failed to parse beta-factor at line 12010 @> WARNING failed to parse beta-factor at line 12011 @> WARNING failed to parse beta-factor at line 12012 @> WARNING failed to parse beta-factor at line 12013 @> WARNING failed to parse beta-factor at line 12014 @> WARNING failed to parse beta-factor at line 12015 @> WARNING failed to parse beta-factor at line 12016 @> WARNING failed to parse beta-factor at line 12017 @> WARNING failed to parse beta-factor at line 12018 @> WARNING failed to parse beta-factor at line 12019 @> WARNING failed to parse beta-factor at line 12020 @> WARNING failed to parse beta-factor at line 12021 @> WARNING failed to parse beta-factor at line 12022 @> WARNING failed to parse beta-factor at line 12023 @> WARNING failed to parse beta-factor at line 12024 @> WARNING failed to parse beta-factor at line 12025 @> WARNING failed to parse beta-factor at line 12026 @> WARNING failed to parse beta-factor at line 12027 @> WARNING failed to parse beta-factor at line 12028 @> WARNING failed to parse beta-factor at line 12029 @> WARNING failed to parse beta-factor at line 12030 @> WARNING failed to parse beta-factor at line 12031 @> WARNING failed to parse beta-factor at line 12032 @> WARNING failed to parse beta-factor at line 12033 @> WARNING failed to parse beta-factor at line 12034 @> WARNING failed to parse beta-factor at line 12035 @> WARNING failed to parse beta-factor at line 12036 @> WARNING failed to parse beta-factor at line 12037 @> WARNING failed to parse beta-factor at line 12038 @> WARNING failed to parse beta-factor at line 12039 @> WARNING failed to parse beta-factor at line 12040 @> WARNING failed to parse beta-factor at line 12041 @> WARNING failed to parse beta-factor at line 12042 @> WARNING failed to parse beta-factor at line 12043 @> WARNING failed to parse beta-factor at line 12044 @> WARNING failed to parse beta-factor at line 12045 @> WARNING failed to parse beta-factor at line 12046 @> WARNING failed to parse beta-factor at line 12047 @> WARNING failed to parse beta-factor at line 12048 @> WARNING failed to parse beta-factor at line 12049 @> WARNING failed to parse beta-factor at line 12050 @> WARNING failed to parse beta-factor at line 12051 @> WARNING failed to parse beta-factor at line 12052 @> WARNING failed to parse beta-factor at line 12053 @> WARNING failed to parse beta-factor at line 12054 @> WARNING failed to parse beta-factor at line 12055 @> WARNING failed to parse beta-factor at line 12056 @> WARNING failed to parse beta-factor at line 12057 @> WARNING failed to parse beta-factor at line 12058 @> WARNING failed to parse beta-factor at line 12059 @> WARNING failed to parse beta-factor at line 12060 @> WARNING failed to parse beta-factor at line 12061 @> WARNING failed to parse beta-factor at line 12062 @> WARNING failed to parse beta-factor at line 12063 @> WARNING failed to parse beta-factor at line 12064 @> WARNING failed to parse beta-factor at line 12065 @> WARNING failed to parse beta-factor at line 12066 @> WARNING failed to parse beta-factor at line 12067 @> WARNING failed to parse beta-factor at line 12068 @> WARNING failed to parse beta-factor at line 12069 @> WARNING failed to parse beta-factor at line 12070 @> WARNING failed to parse beta-factor at line 12071 @> WARNING failed to parse beta-factor at line 12072 @> WARNING failed to parse beta-factor at line 12073 @> WARNING failed to parse beta-factor at line 12074 @> WARNING failed to parse beta-factor at line 12075 @> WARNING failed to parse beta-factor at line 12076 @> WARNING failed to parse beta-factor at line 12077 @> WARNING failed to parse beta-factor at line 12078 @> WARNING failed to parse beta-factor at line 12079 @> WARNING failed to parse beta-factor at line 12080 @> WARNING failed to parse beta-factor at line 12081 @> WARNING failed to parse beta-factor at line 12082 @> WARNING failed to parse beta-factor at line 12083 @> WARNING failed to parse beta-factor at line 12084 @> WARNING failed to parse beta-factor at line 12085 @> WARNING failed to parse beta-factor at line 12086 @> WARNING failed to parse beta-factor at line 12087 @> WARNING failed to parse beta-factor at line 12088 @> WARNING failed to parse beta-factor at line 12089 @> WARNING failed to parse beta-factor at line 12090 @> WARNING failed to parse beta-factor at line 12091 @> WARNING failed to parse beta-factor at line 12092 @> WARNING failed to parse beta-factor at line 12093 @> WARNING failed to parse beta-factor at line 12094 @> WARNING failed to parse beta-factor at line 12095 @> WARNING failed to parse beta-factor at line 12096 @> WARNING failed to parse beta-factor at line 12097 @> WARNING failed to parse beta-factor at line 12098 @> WARNING failed to parse beta-factor at line 12099 @> WARNING failed to parse beta-factor at line 12100 @> WARNING failed to parse beta-factor at line 12101 @> WARNING failed to parse beta-factor at line 12102 @> WARNING failed to parse beta-factor at line 12103 @> WARNING failed to parse beta-factor at line 12104 @> WARNING failed to parse beta-factor at line 12105 @> WARNING failed to parse beta-factor at line 12106 @> WARNING failed to parse beta-factor at line 12107 @> WARNING failed to parse beta-factor at line 12108 @> WARNING failed to parse beta-factor at line 12109 @> WARNING failed to parse beta-factor at line 12110 @> WARNING failed to parse beta-factor at line 12111 @> WARNING failed to parse beta-factor at line 12112 @> WARNING failed to parse beta-factor at line 12113 @> WARNING failed to parse beta-factor at line 12114 @> WARNING failed to parse beta-factor at line 12115 @> WARNING failed to parse beta-factor at line 12116 @> WARNING failed to parse beta-factor at line 12117 @> WARNING failed to parse beta-factor at line 12118 @> WARNING failed to parse beta-factor at line 12119 @> WARNING failed to parse beta-factor at line 12120 @> WARNING failed to parse beta-factor at line 12121 @> WARNING failed to parse beta-factor at line 12122 @> WARNING failed to parse beta-factor at line 12123 @> WARNING failed to parse beta-factor at line 12124 @> WARNING failed to parse beta-factor at line 12125 @> WARNING failed to parse beta-factor at line 12126 @> WARNING failed to parse beta-factor at line 12127 @> WARNING failed to parse beta-factor at line 12128 @> WARNING failed to parse beta-factor at line 12129 @> WARNING failed to parse beta-factor at line 12130 @> WARNING failed to parse beta-factor at line 12131 @> WARNING failed to parse beta-factor at line 12132 @> WARNING failed to parse beta-factor at line 12133 @> WARNING failed to parse beta-factor at line 12134 @> WARNING failed to parse beta-factor at line 12135 @> WARNING failed to parse beta-factor at line 12136 @> WARNING failed to parse beta-factor at line 12137 @> WARNING failed to parse beta-factor at line 12138 @> WARNING failed to parse beta-factor at line 12139 @> WARNING failed to parse beta-factor at line 12140 @> WARNING failed to parse beta-factor at line 12141 @> WARNING failed to parse beta-factor at line 12142 @> WARNING failed to parse beta-factor at line 12143 @> WARNING failed to parse beta-factor at line 12144 @> WARNING failed to parse beta-factor at line 12145 @> WARNING failed to parse beta-factor at line 12146 @> WARNING failed to parse beta-factor at line 12147 @> WARNING failed to parse beta-factor at line 12148 @> WARNING failed to parse beta-factor at line 12149 @> WARNING failed to parse beta-factor at line 12150 @> WARNING failed to parse beta-factor at line 12151 @> WARNING failed to parse beta-factor at line 12152 @> WARNING failed to parse beta-factor at line 12153 @> WARNING failed to parse beta-factor at line 12154 @> WARNING failed to parse beta-factor at line 12155 @> WARNING failed to parse beta-factor at line 12156 @> WARNING failed to parse beta-factor at line 12157 @> WARNING failed to parse beta-factor at line 12158 @> WARNING failed to parse beta-factor at line 12159 @> WARNING failed to parse beta-factor at line 12160 @> WARNING failed to parse beta-factor at line 12161 @> WARNING failed to parse beta-factor at line 12162 @> WARNING failed to parse beta-factor at line 12163 @> WARNING failed to parse beta-factor at line 12164 @> WARNING failed to parse beta-factor at line 12165 @> WARNING failed to parse beta-factor at line 12166 @> WARNING failed to parse beta-factor at line 12167 @> WARNING failed to parse beta-factor at line 12168 @> WARNING failed to parse beta-factor at line 12169 @> WARNING failed to parse beta-factor at line 12170 @> WARNING failed to parse beta-factor at line 12171 @> WARNING failed to parse beta-factor at line 12172 @> WARNING failed to parse beta-factor at line 12173 @> WARNING failed to parse beta-factor at line 12174 @> WARNING failed to parse beta-factor at line 12175 @> WARNING failed to parse beta-factor at line 12176 @> WARNING failed to parse beta-factor at line 12177 @> WARNING failed to parse beta-factor at line 12178 @> WARNING failed to parse beta-factor at line 12179 @> WARNING failed to parse beta-factor at line 12180 @> WARNING failed to parse beta-factor at line 12181 @> WARNING failed to parse beta-factor at line 12182 @> WARNING failed to parse beta-factor at line 12183 @> WARNING failed to parse beta-factor at line 12184 @> WARNING failed to parse beta-factor at line 12185 @> WARNING failed to parse beta-factor at line 12186 @> WARNING failed to parse beta-factor at line 12187 @> WARNING failed to parse beta-factor at line 12188 @> WARNING failed to parse beta-factor at line 12189 @> WARNING failed to parse beta-factor at line 12190 @> WARNING failed to parse beta-factor at line 12191 @> WARNING failed to parse beta-factor at line 12192 @> WARNING failed to parse beta-factor at line 12193 @> WARNING failed to parse beta-factor at line 12194 @> WARNING failed to parse beta-factor at line 12195 @> WARNING failed to parse beta-factor at line 12196 @> WARNING failed to parse beta-factor at line 12197 @> WARNING failed to parse beta-factor at line 12198 @> WARNING failed to parse beta-factor at line 12199 @> WARNING failed to parse beta-factor at line 12200 @> WARNING failed to parse beta-factor at line 12201 @> WARNING failed to parse beta-factor at line 12202 @> WARNING failed to parse beta-factor at line 12203 @> WARNING failed to parse beta-factor at line 12204 @> WARNING failed to parse beta-factor at line 12205 @> WARNING failed to parse beta-factor at line 12206 @> WARNING failed to parse beta-factor at line 12207 @> WARNING failed to parse beta-factor at line 12208 @> WARNING failed to parse beta-factor at line 12209 @> WARNING failed to parse beta-factor at line 12210 @> WARNING failed to parse beta-factor at line 12211 @> WARNING failed to parse beta-factor at line 12212 @> WARNING failed to parse beta-factor at line 12213 @> WARNING failed to parse beta-factor at line 12214 @> WARNING failed to parse beta-factor at line 12215 @> WARNING failed to parse beta-factor at line 12216 @> WARNING failed to parse beta-factor at line 12217 @> WARNING failed to parse beta-factor at line 12218 @> WARNING failed to parse beta-factor at line 12219 @> WARNING failed to parse beta-factor at line 12220 @> WARNING failed to parse beta-factor at line 12221 @> WARNING failed to parse beta-factor at line 12222 @> WARNING failed to parse beta-factor at line 12223 @> WARNING failed to parse beta-factor at line 12224 @> WARNING failed to parse beta-factor at line 12225 @> WARNING failed to parse beta-factor at line 12226 @> WARNING failed to parse beta-factor at line 12227 @> WARNING failed to parse beta-factor at line 12228 @> WARNING failed to parse beta-factor at line 12229 @> WARNING failed to parse beta-factor at line 12230 @> WARNING failed to parse beta-factor at line 12231 @> WARNING failed to parse beta-factor at line 12232 @> WARNING failed to parse beta-factor at line 12233 @> WARNING failed to parse beta-factor at line 12234 @> WARNING failed to parse beta-factor at line 12235 @> WARNING failed to parse beta-factor at line 12236 @> WARNING failed to parse beta-factor at line 12237 @> WARNING failed to parse beta-factor at line 12238 @> WARNING failed to parse beta-factor at line 12239 @> WARNING failed to parse beta-factor at line 12240 @> WARNING failed to parse beta-factor at line 12241 @> WARNING failed to parse beta-factor at line 12242 @> WARNING failed to parse beta-factor at line 12243 @> WARNING failed to parse beta-factor at line 12244 @> WARNING failed to parse beta-factor at line 12245 @> WARNING failed to parse beta-factor at line 12246 @> WARNING failed to parse beta-factor at line 12247 @> WARNING failed to parse beta-factor at line 12248 @> WARNING failed to parse beta-factor at line 12249 @> WARNING failed to parse beta-factor at line 12250 @> WARNING failed to parse beta-factor at line 12251 @> WARNING failed to parse beta-factor at line 12252 @> WARNING failed to parse beta-factor at line 12253 @> WARNING failed to parse beta-factor at line 12254 @> WARNING failed to parse beta-factor at line 12255 @> WARNING failed to parse beta-factor at line 12256 @> WARNING failed to parse beta-factor at line 12257 @> WARNING failed to parse beta-factor at line 12258 @> WARNING failed to parse beta-factor at line 12259 @> WARNING failed to parse beta-factor at line 12260 @> WARNING failed to parse beta-factor at line 12261 @> WARNING failed to parse beta-factor at line 12262 @> WARNING failed to parse beta-factor at line 12263 @> WARNING failed to parse beta-factor at line 12264 @> WARNING failed to parse beta-factor at line 12265 @> WARNING failed to parse beta-factor at line 12266 @> WARNING failed to parse beta-factor at line 12267 @> WARNING failed to parse beta-factor at line 12268 @> WARNING failed to parse beta-factor at line 12269 @> WARNING failed to parse beta-factor at line 12270 @> WARNING failed to parse beta-factor at line 12271 @> WARNING failed to parse beta-factor at line 12272 @> WARNING failed to parse beta-factor at line 12273 @> WARNING failed to parse beta-factor at line 12274 @> WARNING failed to parse beta-factor at line 12275 @> WARNING failed to parse beta-factor at line 12276 @> WARNING failed to parse beta-factor at line 12277 @> WARNING failed to parse beta-factor at line 12278 @> WARNING failed to parse beta-factor at line 12279 @> WARNING failed to parse beta-factor at line 12280 @> WARNING failed to parse beta-factor at line 12281 @> WARNING failed to parse beta-factor at line 12282 @> WARNING failed to parse beta-factor at line 12283 @> WARNING failed to parse beta-factor at line 12284 @> WARNING failed to parse beta-factor at line 12285 @> WARNING failed to parse beta-factor at line 12286 @> WARNING failed to parse beta-factor at line 12287 @> WARNING failed to parse beta-factor at line 12288 @> WARNING failed to parse beta-factor at line 12289 @> WARNING failed to parse beta-factor at line 12290 @> WARNING failed to parse beta-factor at line 12291 @> WARNING failed to parse beta-factor at line 12292 @> WARNING failed to parse beta-factor at line 12293 @> WARNING failed to parse beta-factor at line 12294 @> WARNING failed to parse beta-factor at line 12295 @> WARNING failed to parse beta-factor at line 12296 @> WARNING failed to parse beta-factor at line 12297 @> WARNING failed to parse beta-factor at line 12298 @> WARNING failed to parse beta-factor at line 12299 @> WARNING failed to parse beta-factor at line 12300 @> WARNING failed to parse beta-factor at line 12301 @> WARNING failed to parse beta-factor at line 12302 @> WARNING failed to parse beta-factor at line 12303 @> WARNING failed to parse beta-factor at line 12304 @> WARNING failed to parse beta-factor at line 12305 @> WARNING failed to parse beta-factor at line 12306 @> WARNING failed to parse beta-factor at line 12307 @> WARNING failed to parse beta-factor at line 12308 @> WARNING failed to parse beta-factor at line 12309 @> WARNING failed to parse beta-factor at line 12310 @> WARNING failed to parse beta-factor at line 12311 @> WARNING failed to parse beta-factor at line 12312 @> WARNING failed to parse beta-factor at line 12313 @> WARNING failed to parse beta-factor at line 12314 @> WARNING failed to parse beta-factor at line 12315 @> WARNING failed to parse beta-factor at line 12316 @> WARNING failed to parse beta-factor at line 12317 @> WARNING failed to parse beta-factor at line 12318 @> WARNING failed to parse beta-factor at line 12319 @> WARNING failed to parse beta-factor at line 12320 @> WARNING failed to parse beta-factor at line 12321 @> WARNING failed to parse beta-factor at line 12322 @> WARNING failed to parse beta-factor at line 12323 @> WARNING failed to parse beta-factor at line 12324 @> WARNING failed to parse beta-factor at line 12325 @> WARNING failed to parse beta-factor at line 12326 @> WARNING failed to parse beta-factor at line 12327 @> WARNING failed to parse beta-factor at line 12328 @> WARNING failed to parse beta-factor at line 12329 @> WARNING failed to parse beta-factor at line 12330 @> WARNING failed to parse beta-factor at line 12331 @> WARNING failed to parse beta-factor at line 12332 @> WARNING failed to parse beta-factor at line 12333 @> WARNING failed to parse beta-factor at line 12334 @> WARNING failed to parse beta-factor at line 12335 @> WARNING failed to parse beta-factor at line 12336 @> WARNING failed to parse beta-factor at line 12337 @> WARNING failed to parse beta-factor at line 12338 @> WARNING failed to parse beta-factor at line 12339 @> WARNING failed to parse beta-factor at line 12340 @> WARNING failed to parse beta-factor at line 12341 @> WARNING failed to parse beta-factor at line 12342 @> WARNING failed to parse beta-factor at line 12343 @> WARNING failed to parse beta-factor at line 12344 @> WARNING failed to parse beta-factor at line 12345 @> WARNING failed to parse beta-factor at line 12346 @> WARNING failed to parse beta-factor at line 12347 @> WARNING failed to parse beta-factor at line 12348 @> WARNING failed to parse beta-factor at line 12349 @> WARNING failed to parse beta-factor at line 12350 @> WARNING failed to parse beta-factor at line 12351 @> WARNING failed to parse beta-factor at line 12352 @> WARNING failed to parse beta-factor at line 12353 @> WARNING failed to parse beta-factor at line 12354 @> WARNING failed to parse beta-factor at line 12355 @> WARNING failed to parse beta-factor at line 12356 @> WARNING failed to parse beta-factor at line 12357 @> WARNING failed to parse beta-factor at line 12358 @> WARNING failed to parse beta-factor at line 12359 @> WARNING failed to parse beta-factor at line 12360 @> WARNING failed to parse beta-factor at line 12361 @> WARNING failed to parse beta-factor at line 12362 @> WARNING failed to parse beta-factor at line 12363 @> WARNING failed to parse beta-factor at line 12364 @> WARNING failed to parse beta-factor at line 12365 @> WARNING failed to parse beta-factor at line 12366 @> WARNING failed to parse beta-factor at line 12367 @> WARNING failed to parse beta-factor at line 12368 @> WARNING failed to parse beta-factor at line 12369 @> WARNING failed to parse beta-factor at line 12370 @> WARNING failed to parse beta-factor at line 12371 @> WARNING failed to parse beta-factor at line 12372 @> WARNING failed to parse beta-factor at line 12373 @> WARNING failed to parse beta-factor at line 12374 @> WARNING failed to parse beta-factor at line 12375 @> WARNING failed to parse beta-factor at line 12376 @> WARNING failed to parse beta-factor at line 12377 @> WARNING failed to parse beta-factor at line 12378 @> WARNING failed to parse beta-factor at line 12379 @> WARNING failed to parse beta-factor at line 12380 @> WARNING failed to parse beta-factor at line 12381 @> WARNING failed to parse beta-factor at line 12382 @> WARNING failed to parse beta-factor at line 12383 @> WARNING failed to parse beta-factor at line 12384 @> WARNING failed to parse beta-factor at line 12385 @> WARNING failed to parse beta-factor at line 12386 @> WARNING failed to parse beta-factor at line 12387 @> WARNING failed to parse beta-factor at line 12388 @> WARNING failed to parse beta-factor at line 12389 @> WARNING failed to parse beta-factor at line 12390 @> WARNING failed to parse beta-factor at line 12391 @> WARNING failed to parse beta-factor at line 12392 @> WARNING failed to parse beta-factor at line 12393 @> WARNING failed to parse beta-factor at line 12394 @> WARNING failed to parse beta-factor at line 12395 @> WARNING failed to parse beta-factor at line 12396 @> WARNING failed to parse beta-factor at line 12397 @> WARNING failed to parse beta-factor at line 12398 @> WARNING failed to parse beta-factor at line 12399 @> WARNING failed to parse beta-factor at line 12400 @> WARNING failed to parse beta-factor at line 12401 @> WARNING failed to parse beta-factor at line 12402 @> WARNING failed to parse beta-factor at line 12403 @> WARNING failed to parse beta-factor at line 12404 @> WARNING failed to parse beta-factor at line 12405 @> WARNING failed to parse beta-factor at line 12406 @> WARNING failed to parse beta-factor at line 12407 @> WARNING failed to parse beta-factor at line 12408 @> WARNING failed to parse beta-factor at line 12409 @> WARNING failed to parse beta-factor at line 12410 @> WARNING failed to parse beta-factor at line 12411 @> WARNING failed to parse beta-factor at line 12412 @> WARNING failed to parse beta-factor at line 12413 @> WARNING failed to parse beta-factor at line 12414 @> WARNING failed to parse beta-factor at line 12415 @> WARNING failed to parse beta-factor at line 12416 @> WARNING failed to parse beta-factor at line 12417 @> WARNING failed to parse beta-factor at line 12418 @> WARNING failed to parse beta-factor at line 12419 @> WARNING failed to parse beta-factor at line 12420 @> WARNING failed to parse beta-factor at line 12421 @> WARNING failed to parse beta-factor at line 12422 @> WARNING failed to parse beta-factor at line 12423 @> WARNING failed to parse beta-factor at line 12424 @> WARNING failed to parse beta-factor at line 12425 @> WARNING failed to parse beta-factor at line 12426 @> WARNING failed to parse beta-factor at line 12427 @> WARNING failed to parse beta-factor at line 12428 @> WARNING failed to parse beta-factor at line 12429 @> WARNING failed to parse beta-factor at line 12430 @> WARNING failed to parse beta-factor at line 12431 @> WARNING failed to parse beta-factor at line 12432 @> WARNING failed to parse beta-factor at line 12433 @> WARNING failed to parse beta-factor at line 12434 @> WARNING failed to parse beta-factor at line 12435 @> WARNING failed to parse beta-factor at line 12436 @> WARNING failed to parse beta-factor at line 12437 @> WARNING failed to parse beta-factor at line 12438 @> WARNING failed to parse beta-factor at line 12439 @> WARNING failed to parse beta-factor at line 12440 @> WARNING failed to parse beta-factor at line 12441 @> WARNING failed to parse beta-factor at line 12442 @> WARNING failed to parse beta-factor at line 12443 @> WARNING failed to parse beta-factor at line 12444 @> WARNING failed to parse beta-factor at line 12445 @> WARNING failed to parse beta-factor at line 12446 @> WARNING failed to parse beta-factor at line 12447 @> WARNING failed to parse beta-factor at line 12448 @> WARNING failed to parse beta-factor at line 12449 @> WARNING failed to parse beta-factor at line 12450 @> WARNING failed to parse beta-factor at line 12451 @> WARNING failed to parse beta-factor at line 12452 @> WARNING failed to parse beta-factor at line 12453 @> WARNING failed to parse beta-factor at line 12454 @> WARNING failed to parse beta-factor at line 12455 @> WARNING failed to parse beta-factor at line 12456 @> WARNING failed to parse beta-factor at line 12457 @> WARNING failed to parse beta-factor at line 12458 @> WARNING failed to parse beta-factor at line 12459 @> WARNING failed to parse beta-factor at line 12460 @> WARNING failed to parse beta-factor at line 12461 @> WARNING failed to parse beta-factor at line 12462 @> WARNING failed to parse beta-factor at line 12463 @> WARNING failed to parse beta-factor at line 12464 @> WARNING failed to parse beta-factor at line 12465 @> WARNING failed to parse beta-factor at line 12466 @> WARNING failed to parse beta-factor at line 12467 @> WARNING failed to parse beta-factor at line 12468 @> WARNING failed to parse beta-factor at line 12469 @> WARNING failed to parse beta-factor at line 12470 @> WARNING failed to parse beta-factor at line 12471 @> WARNING failed to parse beta-factor at line 12472 @> WARNING failed to parse beta-factor at line 12473 @> WARNING failed to parse beta-factor at line 12474 @> WARNING failed to parse beta-factor at line 12475 @> WARNING failed to parse beta-factor at line 12476 @> WARNING failed to parse beta-factor at line 12477 @> WARNING failed to parse beta-factor at line 12478 @> WARNING failed to parse beta-factor at line 12479 @> WARNING failed to parse beta-factor at line 12480 @> WARNING failed to parse beta-factor at line 12481 @> WARNING failed to parse beta-factor at line 12482 @> WARNING failed to parse beta-factor at line 12483 @> WARNING failed to parse beta-factor at line 12484 @> WARNING failed to parse beta-factor at line 12485 @> WARNING failed to parse beta-factor at line 12486 @> WARNING failed to parse beta-factor at line 12487 @> WARNING failed to parse beta-factor at line 12488 @> WARNING failed to parse beta-factor at line 12489 @> WARNING failed to parse beta-factor at line 12490 @> WARNING failed to parse beta-factor at line 12491 @> WARNING failed to parse beta-factor at line 12492 @> WARNING failed to parse beta-factor at line 12493 @> WARNING failed to parse beta-factor at line 12494 @> WARNING failed to parse beta-factor at line 12495 @> WARNING failed to parse beta-factor at line 12496 @> WARNING failed to parse beta-factor at line 12497 @> WARNING failed to parse beta-factor at line 12498 @> WARNING failed to parse beta-factor at line 12499 @> WARNING failed to parse beta-factor at line 12500 @> WARNING failed to parse beta-factor at line 12501 @> WARNING failed to parse beta-factor at line 12502 @> WARNING failed to parse beta-factor at line 12503 @> WARNING failed to parse beta-factor at line 12504 @> WARNING failed to parse beta-factor at line 12505 @> WARNING failed to parse beta-factor at line 12506 @> WARNING failed to parse beta-factor at line 12507 @> WARNING failed to parse beta-factor at line 12508 @> WARNING failed to parse beta-factor at line 12509 @> WARNING failed to parse beta-factor at line 12510 @> WARNING failed to parse beta-factor at line 12511 @> WARNING failed to parse beta-factor at line 12512 @> WARNING failed to parse beta-factor at line 12513 @> WARNING failed to parse beta-factor at line 12514 @> WARNING failed to parse beta-factor at line 12515 @> WARNING failed to parse beta-factor at line 12516 @> WARNING failed to parse beta-factor at line 12517 @> WARNING failed to parse beta-factor at line 12518 @> WARNING failed to parse beta-factor at line 12519 @> WARNING failed to parse beta-factor at line 12520 @> WARNING failed to parse beta-factor at line 12521 @> WARNING failed to parse beta-factor at line 12522 @> WARNING failed to parse beta-factor at line 12523 @> WARNING failed to parse beta-factor at line 12524 @> WARNING failed to parse beta-factor at line 12525 @> WARNING failed to parse beta-factor at line 12526 @> WARNING failed to parse beta-factor at line 12527 @> WARNING failed to parse beta-factor at line 12528 @> WARNING failed to parse beta-factor at line 12529 @> WARNING failed to parse beta-factor at line 12530 @> WARNING failed to parse beta-factor at line 12531 @> WARNING failed to parse beta-factor at line 12532 @> WARNING failed to parse beta-factor at line 12533 @> WARNING failed to parse beta-factor at line 12534 @> WARNING failed to parse beta-factor at line 12535 @> WARNING failed to parse beta-factor at line 12536 @> WARNING failed to parse beta-factor at line 12537 @> WARNING failed to parse beta-factor at line 12538 @> WARNING failed to parse beta-factor at line 12539 @> WARNING failed to parse beta-factor at line 12540 @> WARNING failed to parse beta-factor at line 12541 @> WARNING failed to parse beta-factor at line 12542 @> WARNING failed to parse beta-factor at line 12543 @> WARNING failed to parse beta-factor at line 12544 @> WARNING failed to parse beta-factor at line 12545 @> WARNING failed to parse beta-factor at line 12546 @> WARNING failed to parse beta-factor at line 12547 @> WARNING failed to parse beta-factor at line 12548 @> WARNING failed to parse beta-factor at line 12549 @> WARNING failed to parse beta-factor at line 12550 @> WARNING failed to parse beta-factor at line 12551 @> WARNING failed to parse beta-factor at line 12552 @> WARNING failed to parse beta-factor at line 12553 @> WARNING failed to parse beta-factor at line 12554 @> WARNING failed to parse beta-factor at line 12555 @> WARNING failed to parse beta-factor at line 12556 @> WARNING failed to parse beta-factor at line 12557 @> WARNING failed to parse beta-factor at line 12558 @> WARNING failed to parse beta-factor at line 12559 @> WARNING failed to parse beta-factor at line 12560 @> WARNING failed to parse beta-factor at line 12561 @> WARNING failed to parse beta-factor at line 12562 @> WARNING failed to parse beta-factor at line 12563 @> WARNING failed to parse beta-factor at line 12564 @> WARNING failed to parse beta-factor at line 12565 @> WARNING failed to parse beta-factor at line 12566 @> WARNING failed to parse beta-factor at line 12567 @> WARNING failed to parse beta-factor at line 12568 @> WARNING failed to parse beta-factor at line 12569 @> WARNING failed to parse beta-factor at line 12570 @> WARNING failed to parse beta-factor at line 12571 @> WARNING failed to parse beta-factor at line 12572 @> WARNING failed to parse beta-factor at line 12573 @> WARNING failed to parse beta-factor at line 12574 @> WARNING failed to parse beta-factor at line 12575 @> WARNING failed to parse beta-factor at line 12576 @> WARNING failed to parse beta-factor at line 12577 @> WARNING failed to parse beta-factor at line 12578 @> WARNING failed to parse beta-factor at line 12579 @> WARNING failed to parse beta-factor at line 12580 @> WARNING failed to parse beta-factor at line 12581 @> WARNING failed to parse beta-factor at line 12582 @> WARNING failed to parse beta-factor at line 12583 @> WARNING failed to parse beta-factor at line 12584 @> WARNING failed to parse beta-factor at line 12585 @> WARNING failed to parse beta-factor at line 12586 @> WARNING failed to parse beta-factor at line 12587 @> WARNING failed to parse beta-factor at line 12588 @> WARNING failed to parse beta-factor at line 12589 @> WARNING failed to parse beta-factor at line 12590 @> WARNING failed to parse beta-factor at line 12591 @> WARNING failed to parse beta-factor at line 12592 @> WARNING failed to parse beta-factor at line 12593 @> WARNING failed to parse beta-factor at line 12594 @> WARNING failed to parse beta-factor at line 12595 @> WARNING failed to parse beta-factor at line 12596 @> WARNING failed to parse beta-factor at line 12597 @> WARNING failed to parse beta-factor at line 12598 @> WARNING failed to parse beta-factor at line 12599 @> WARNING failed to parse beta-factor at line 12600 @> WARNING failed to parse beta-factor at line 12601 @> WARNING failed to parse beta-factor at line 12602 @> WARNING failed to parse beta-factor at line 12603 @> WARNING failed to parse beta-factor at line 12604 @> WARNING failed to parse beta-factor at line 12605 @> WARNING failed to parse beta-factor at line 12606 @> WARNING failed to parse beta-factor at line 12607 @> WARNING failed to parse beta-factor at line 12608 @> WARNING failed to parse beta-factor at line 12609 @> WARNING failed to parse beta-factor at line 12610 @> WARNING failed to parse beta-factor at line 12611 @> WARNING failed to parse beta-factor at line 12612 @> WARNING failed to parse beta-factor at line 12613 @> WARNING failed to parse beta-factor at line 12614 @> WARNING failed to parse beta-factor at line 12615 @> WARNING failed to parse beta-factor at line 12616 @> WARNING failed to parse beta-factor at line 12617 @> WARNING failed to parse beta-factor at line 12618 @> WARNING failed to parse beta-factor at line 12619 @> WARNING failed to parse beta-factor at line 12620 @> WARNING failed to parse beta-factor at line 12621 @> WARNING failed to parse beta-factor at line 12622 @> WARNING failed to parse beta-factor at line 12623 @> WARNING failed to parse beta-factor at line 12624 @> WARNING failed to parse beta-factor at line 12625 @> WARNING failed to parse beta-factor at line 12626 @> WARNING failed to parse beta-factor at line 12627 IOPub message rate exceeded. The notebook server will temporarily stop sending output to the client in order to avoid crashing it. To change this limit, set the config variable `--NotebookApp.iopub_msg_rate_limit`. Current values: NotebookApp.iopub_msg_rate_limit=1000.0 (msgs/sec) NotebookApp.rate_limit_window=3.0 (secs)
There will be warnings saying that ProDy wants to read beta factors, but the coordinates should be read properly. In addition to atoms, the OPM file contains points to indicate the boundaries of the membrane.
We now make two selections, one from each structure. The selections are chosen so that the final structures are homotrimers with an equal number (398) of atoms in each subunit. We also want to remove the three aspartate ligands, which are indicated as chain D in the outward-facing structure and have resid 500, in the inward-facing structure.
of_ca = of_all.select('protein and name CA and not (chain A and resid 119 to 122) and not (chain C and resid 119 to 123) and not chain D')
if_ca = if_all.select('protein and name CA and not (resid 6 to 9) and not (resid 119 to 127) and resid < 500')
As a last step in preparation, we can align the structures so that we can calculate a deformation vector and compare the modes to it, as shown in the ENM Tutorial.
superpose(if_ca, of_ca)
(<Selection: 'protein and nam...and resid < 500' from 3KBC-opm (1194 atoms)>, <prody.measure.transform.Transformation at 0x19b366a60>)
ProDy’s RTB method can be used for any system, whether or not a membrane is involved. imANM is an extension of RTB and they are in the same part of ProDy. The RTB method allows us to decompose the protein into pre-defined rigid blocks. Atoms within a block do not move relative to each other (hence the descriptor “rigid”), but blocks can move relative to other blocks. There are two main benefits of using blocks: First, the Hessian for a good blocking scheme is smaller than the Hessian for an all-residue representation, so the modes can be calculated more quickly. This is particularly useful when one is considering very large systems (in this case, containing thousands of residues). Second, the use of rigid blocks reduces unphysical distortions of the structure, such as stretching of backbone bonds that may result from the harmonic approximation. These benefits come at the price of accuracy. Imposing rigidity reduces the amount of dynamical detail that can be recovered from the model.
In ProDy, a rigid block is defined as a set of atoms that move together (i.e., the distances between them are fixed). Typically the constituent atoms of a rigid block are spatially adjacent (i.e., they all belong to the same domain or secondary structure element), but users are free to define blocks however they wish. The only restriction is that a block cannot contain exactly two particles. This restriction is in place because it is mathematically inconvenient to deal with two-particle blocks.
We can either define blocks within our python session, or define them externally in a separate file and write a little bit of code to handle the tasks of reading the file and assigning residues to blocks. This latter approach can be useful when exploring and comparing many different blocking schemes. We have developed one such format for a block file, examples of which can be found in 2nwl_blocks.txt and 3kbc_blocks.txt. The first ten lines of 2nwl_blocks.txt are:
The columns, separated by whitespace, are formatted as follows:
This is just one way of storing information on how the protein is deconstructed into blocks. You are welcome to use others if you have a way of reading them. We can read blocks from 2nwl_blocks.txt into the array blocks as follows:
def set_block(filename, ag):
ag.setData('block', 0)
with open(filename) as inp:
for line in inp:
b, n1, c1, r1, n2, c2, r2 = line.split()
sel = ag.select('chain {} and resnum {} to {}'
.format(c1, r1, r2))
if sel != None:
sel.setData('block', b)
set_block('./membrane_anm_files/2nwl_blocks.txt', of_ca.getAtomGroup())
of_blocks = of_ca.getData('block')
blk='./membrane_anm_files/2nwl_blocks.txt'
of_ca.getAtomGroup().setData('block', 0)
with open(blk) as inp:
for line in inp:
b, n1, c1, r1, n2, c2, r2 = line.split()
sel = of_ca.select('chain {} and resnum {} to {}'
.format(c1, r1, r2))
if sel != None:
sel.setData('block', b)
of_blocks = of_ca.getData('block')
of_blocks
array([ 1, 1, 1, ..., 263, 263, 263])
We will do the same for the blocks of the inward-facing structure. The block definitions are based on secondary structures, which vary slightly between the structures. We therefore have two separate blocking schemes.
set_block('./membrane_anm_files/3kbc_blocks.txt', if_ca.getAtomGroup())
if_blocks = if_ca.getData('block')
if_blocks
array([ 1, 2, 3, ..., 260, 260, 261])
To use the blocks in an RTB ANM calculation, we instantiate an RTB object for each structure:
of_rtb = RTB('2nwl')
if_rtb = RTB('3kbc')
and we build a couple of Hessians using the coordinates of the crystal structures
of_coords = of_ca.getCoords()
of_rtb.buildHessian(of_coords, of_blocks, cutoff=11.0, scale=16., membrane_low=-1000.0, membrane_high=1000.0)
@> Hessian was built in 0.32s. @> System has 233 blocks, the largest with 29 of 1194 units. @> Block Hessian and projection matrix were calculated in 0.37s.
if_coords = if_ca.getCoords()
if_rtb.buildHessian(if_coords, if_blocks, cutoff=11.0, scale=16., membrane_low=-1000.0, membrane_high=1000.0)
@> Hessian was built in 0.32s. @> System has 249 blocks, the largest with 29 of 1194 units. @> Block Hessian and projection matrix were calculated in 0.14s.
The scaling factor of 16 in this example means that the restoring force for any displacement in the x- or y-direction is 16 times greater than the force associated with a displacement in the z-direction. The constraint on motions parallel to the membrane surface implicitly incorporates the membrane’s effects into ANM. To use RTB with no membrane effects, set scale=1.0 (which is also the default value). We have here set the boundaries of the membrane to extend well beyond the protein, effectively applying the implicit membrane scaling to the entire protein.
Now we calculate the modes and write them to a pair of .nmd files for viewing.
of_rtb.calcModes()
@> 20 modes were calculated in 0.12s.
if_rtb.calcModes()
@> 20 modes were calculated in 0.09s.
writeNMD('2nwl_im.nmd', of_rtb, of_ca.select('protein and name CA'))
'2nwl_im.nmd'
writeNMD('3kbc_im.nmd', if_rtb, if_ca.select('protein and name CA'))
'3kbc_im.nmd'
The third mode of the outward-facing structure moves all three transport domains simultaneously through the membrane in a 'lift-like' motion.
A similar motion is shown in mode 6 of the inward-facing structure.
As in the previous section, we will investigate the effect of the presence of membrane for the motion of a neurotransmitter transporter with ProDy's explicit membrane ANM (exANM) capabilities. The procedure relies on explicitly building a membrane lattice and using reduceModel().
The exANM assumes that the membrane is normal to the z-axis, so it is important to use a structure that is properly aligned. The structure from the OPM database will work.
of_all = parsePDB('./membrane_anm_files/2NWL-opm.pdb') # Outward-facing structure
@> WARNING failed to parse beta-factor at line 8730 @> WARNING failed to parse beta-factor at line 8731 @> WARNING failed to parse beta-factor at line 8732 @> WARNING failed to parse beta-factor at line 8733 @> WARNING failed to parse beta-factor at line 8734 @> WARNING failed to parse beta-factor at line 8735 @> WARNING failed to parse beta-factor at line 8736 @> WARNING failed to parse beta-factor at line 8737 @> WARNING failed to parse beta-factor at line 8738 @> WARNING failed to parse beta-factor at line 8739 @> WARNING failed to parse beta-factor at line 8740 @> WARNING failed to parse beta-factor at line 8741 @> WARNING failed to parse beta-factor at line 8742 @> WARNING failed to parse beta-factor at line 8743 @> WARNING failed to parse beta-factor at line 8744 @> WARNING failed to parse beta-factor at line 8745 @> WARNING failed to parse beta-factor at line 8746 @> WARNING failed to parse beta-factor at line 8747 @> WARNING failed to parse beta-factor at line 8748 @> WARNING failed to parse beta-factor at line 8749 @> WARNING failed to parse beta-factor at line 8750 @> WARNING failed to parse beta-factor at line 8751 @> WARNING failed to parse beta-factor at line 8752 @> WARNING failed to parse beta-factor at line 8753 @> WARNING failed to parse beta-factor at line 8754 @> WARNING failed to parse beta-factor at line 8755 @> WARNING failed to parse beta-factor at line 8756 @> WARNING failed to parse beta-factor at line 8757 @> WARNING failed to parse beta-factor at line 8758 @> WARNING failed to parse beta-factor at line 8759 @> WARNING failed to parse beta-factor at line 8760 @> WARNING failed to parse beta-factor at line 8761 @> WARNING failed to parse beta-factor at line 8762 @> WARNING failed to parse beta-factor at line 8763 @> WARNING failed to parse beta-factor at line 8764 @> WARNING failed to parse beta-factor at line 8765 @> WARNING failed to parse beta-factor at line 8766 @> WARNING failed to parse beta-factor at line 8767 @> WARNING failed to parse beta-factor at line 8768 @> WARNING failed to parse beta-factor at line 8769 @> WARNING failed to parse beta-factor at line 8770 @> WARNING failed to parse beta-factor at line 8771 @> WARNING failed to parse beta-factor at line 8772 @> WARNING failed to parse beta-factor at line 8773 @> WARNING failed to parse beta-factor at line 8774 @> WARNING failed to parse beta-factor at line 8775 @> WARNING failed to parse beta-factor at line 8776 @> WARNING failed to parse beta-factor at line 8777 @> WARNING failed to parse beta-factor at line 8778 @> WARNING failed to parse beta-factor at line 8779 @> WARNING failed to parse beta-factor at line 8780 @> WARNING failed to parse beta-factor at line 8781 @> WARNING failed to parse beta-factor at line 8782 @> WARNING failed to parse beta-factor at line 8783 @> WARNING failed to parse beta-factor at line 8784 @> WARNING failed to parse beta-factor at line 8785 @> WARNING failed to parse beta-factor at line 8786 @> WARNING failed to parse beta-factor at line 8787 @> WARNING failed to parse beta-factor at line 8788 @> WARNING failed to parse beta-factor at line 8789 @> WARNING failed to parse beta-factor at line 8790 @> WARNING failed to parse beta-factor at line 8791 @> WARNING failed to parse beta-factor at line 8792 @> WARNING failed to parse beta-factor at line 8793 @> WARNING failed to parse beta-factor at line 8794 @> WARNING failed to parse beta-factor at line 8795 @> WARNING failed to parse beta-factor at line 8796 @> WARNING failed to parse beta-factor at line 8797 @> WARNING failed to parse beta-factor at line 8798 @> WARNING failed to parse beta-factor at line 8799 @> WARNING failed to parse beta-factor at line 8800 @> WARNING failed to parse beta-factor at line 8801 @> WARNING failed to parse beta-factor at line 8802 @> WARNING failed to parse beta-factor at line 8803 @> WARNING failed to parse beta-factor at line 8804 @> WARNING failed to parse beta-factor at line 8805 @> WARNING failed to parse beta-factor at line 8806 @> WARNING failed to parse beta-factor at line 8807 @> WARNING failed to parse beta-factor at line 8808 @> WARNING failed to parse beta-factor at line 8809 @> WARNING failed to parse beta-factor at line 8810 @> WARNING failed to parse beta-factor at line 8811 @> WARNING failed to parse beta-factor at line 8812 @> WARNING failed to parse beta-factor at line 8813 @> WARNING failed to parse beta-factor at line 8814 @> WARNING failed to parse beta-factor at line 8815 @> WARNING failed to parse beta-factor at line 8816 @> WARNING failed to parse beta-factor at line 8817 @> WARNING failed to parse beta-factor at line 8818 @> WARNING failed to parse beta-factor at line 8819 @> WARNING failed to parse beta-factor at line 8820 @> WARNING failed to parse beta-factor at line 8821 @> WARNING failed to parse beta-factor at line 8822 @> WARNING failed to parse beta-factor at line 8823 @> WARNING failed to parse beta-factor at line 8824 @> WARNING failed to parse beta-factor at line 8825 @> WARNING failed to parse beta-factor at line 8826 @> WARNING failed to parse beta-factor at line 8827 @> WARNING failed to parse beta-factor at line 8828 @> WARNING failed to parse beta-factor at line 8829 @> WARNING failed to parse beta-factor at line 8830 @> WARNING failed to parse beta-factor at line 8831 @> WARNING failed to parse beta-factor at line 8832 @> WARNING failed to parse beta-factor at line 8833 @> WARNING failed to parse beta-factor at line 8834 @> WARNING failed to parse beta-factor at line 8835 @> WARNING failed to parse beta-factor at line 8836 @> WARNING failed to parse beta-factor at line 8837 @> WARNING failed to parse beta-factor at line 8838 @> WARNING failed to parse beta-factor at line 8839 @> WARNING failed to parse beta-factor at line 8840 @> WARNING failed to parse beta-factor at line 8841 @> WARNING failed to parse beta-factor at line 8842 @> WARNING failed to parse beta-factor at line 8843 @> WARNING failed to parse beta-factor at line 8844 @> WARNING failed to parse beta-factor at line 8845 @> WARNING failed to parse beta-factor at line 8846 @> WARNING failed to parse beta-factor at line 8847 @> WARNING failed to parse beta-factor at line 8848 @> WARNING failed to parse beta-factor at line 8849 @> WARNING failed to parse beta-factor at line 8850 @> WARNING failed to parse beta-factor at line 8851 @> WARNING failed to parse beta-factor at line 8852 @> WARNING failed to parse beta-factor at line 8853 @> WARNING failed to parse beta-factor at line 8854 @> WARNING failed to parse beta-factor at line 8855 @> WARNING failed to parse beta-factor at line 8856 @> WARNING failed to parse beta-factor at line 8857 @> WARNING failed to parse beta-factor at line 8858 @> WARNING failed to parse beta-factor at line 8859 @> WARNING failed to parse beta-factor at line 8860 @> WARNING failed to parse beta-factor at line 8861 @> WARNING failed to parse beta-factor at line 8862 @> WARNING failed to parse beta-factor at line 8863 @> WARNING failed to parse beta-factor at line 8864 @> WARNING failed to parse beta-factor at line 8865 @> WARNING failed to parse beta-factor at line 8866 @> WARNING failed to parse beta-factor at line 8867 @> WARNING failed to parse beta-factor at line 8868 @> WARNING failed to parse beta-factor at line 8869 @> WARNING failed to parse beta-factor at line 8870 @> WARNING failed to parse beta-factor at line 8871 @> WARNING failed to parse beta-factor at line 8872 @> WARNING failed to parse beta-factor at line 8873 @> WARNING failed to parse beta-factor at line 8874 @> WARNING failed to parse beta-factor at line 8875 @> WARNING failed to parse beta-factor at line 8876 @> WARNING failed to parse beta-factor at line 8877 @> WARNING failed to parse beta-factor at line 8878 @> WARNING failed to parse beta-factor at line 8879 @> WARNING failed to parse beta-factor at line 8880 @> WARNING failed to parse beta-factor at line 8881 @> WARNING failed to parse beta-factor at line 8882 @> WARNING failed to parse beta-factor at line 8883 @> WARNING failed to parse beta-factor at line 8884 @> WARNING failed to parse beta-factor at line 8885 @> WARNING failed to parse beta-factor at line 8886 @> WARNING failed to parse beta-factor at line 8887 @> WARNING failed to parse beta-factor at line 8888 @> WARNING failed to parse beta-factor at line 8889 @> WARNING failed to parse beta-factor at line 8890 @> WARNING failed to parse beta-factor at line 8891 @> WARNING failed to parse beta-factor at line 8892 @> WARNING failed to parse beta-factor at line 8893 @> WARNING failed to parse beta-factor at line 8894 @> WARNING failed to parse beta-factor at line 8895 @> WARNING failed to parse beta-factor at line 8896 @> WARNING failed to parse beta-factor at line 8897 @> WARNING failed to parse beta-factor at line 8898 @> WARNING failed to parse beta-factor at line 8899 @> WARNING failed to parse beta-factor at line 8900 @> WARNING failed to parse beta-factor at line 8901 @> WARNING failed to parse beta-factor at line 8902 @> WARNING failed to parse beta-factor at line 8903 @> WARNING failed to parse beta-factor at line 8904 @> WARNING failed to parse beta-factor at line 8905 @> WARNING failed to parse beta-factor at line 8906 @> WARNING failed to parse beta-factor at line 8907 @> WARNING failed to parse beta-factor at line 8908 @> WARNING failed to parse beta-factor at line 8909 @> WARNING failed to parse beta-factor at line 8910 @> WARNING failed to parse beta-factor at line 8911 @> WARNING failed to parse beta-factor at line 8912 @> WARNING failed to parse beta-factor at line 8913 @> WARNING failed to parse beta-factor at line 8914 @> WARNING failed to parse beta-factor at line 8915 @> WARNING failed to parse beta-factor at line 8916 @> WARNING failed to parse beta-factor at line 8917 @> WARNING failed to parse beta-factor at line 8918 @> WARNING failed to parse beta-factor at line 8919 @> WARNING failed to parse beta-factor at line 8920 @> WARNING failed to parse beta-factor at line 8921 @> WARNING failed to parse beta-factor at line 8922 @> WARNING failed to parse beta-factor at line 8923 @> WARNING failed to parse beta-factor at line 8924 @> WARNING failed to parse beta-factor at line 8925 @> WARNING failed to parse beta-factor at line 8926 @> WARNING failed to parse beta-factor at line 8927 @> WARNING failed to parse beta-factor at line 8928 @> WARNING failed to parse beta-factor at line 8929 @> WARNING failed to parse beta-factor at line 8930 @> WARNING failed to parse beta-factor at line 8931 @> WARNING failed to parse beta-factor at line 8932 @> WARNING failed to parse beta-factor at line 8933 @> WARNING failed to parse beta-factor at line 8934 @> WARNING failed to parse beta-factor at line 8935 @> WARNING failed to parse beta-factor at line 8936 @> WARNING failed to parse beta-factor at line 8937 @> WARNING failed to parse beta-factor at line 8938 @> WARNING failed to parse beta-factor at line 8939 @> WARNING failed to parse beta-factor at line 8940 @> WARNING failed to parse beta-factor at line 8941 @> WARNING failed to parse beta-factor at line 8942 @> WARNING failed to parse beta-factor at line 8943 @> WARNING failed to parse beta-factor at line 8944 @> WARNING failed to parse beta-factor at line 8945 @> WARNING failed to parse beta-factor at line 8946 @> WARNING failed to parse beta-factor at line 8947 @> WARNING failed to parse beta-factor at line 8948 @> WARNING failed to parse beta-factor at line 8949 @> WARNING failed to parse beta-factor at line 8950 @> WARNING failed to parse beta-factor at line 8951 @> WARNING failed to parse beta-factor at line 8952 @> WARNING failed to parse beta-factor at line 8953 @> WARNING failed to parse beta-factor at line 8954 @> WARNING failed to parse beta-factor at line 8955 @> WARNING failed to parse beta-factor at line 8956 @> WARNING failed to parse beta-factor at line 8957 @> WARNING failed to parse beta-factor at line 8958 @> WARNING failed to parse beta-factor at line 8959 @> WARNING failed to parse beta-factor at line 8960 @> WARNING failed to parse beta-factor at line 8961 @> WARNING failed to parse beta-factor at line 8962 @> WARNING failed to parse beta-factor at line 8963 @> WARNING failed to parse beta-factor at line 8964 @> WARNING failed to parse beta-factor at line 8965 @> WARNING failed to parse beta-factor at line 8966 @> WARNING failed to parse beta-factor at line 8967 @> WARNING failed to parse beta-factor at line 8968 @> WARNING failed to parse beta-factor at line 8969 @> WARNING failed to parse beta-factor at line 8970 @> WARNING failed to parse beta-factor at line 8971 @> WARNING failed to parse beta-factor at line 8972 @> WARNING failed to parse beta-factor at line 8973 @> WARNING failed to parse beta-factor at line 8974 @> WARNING failed to parse beta-factor at line 8975 @> WARNING failed to parse beta-factor at line 8976 @> WARNING failed to parse beta-factor at line 8977 @> WARNING failed to parse beta-factor at line 8978 @> WARNING failed to parse beta-factor at line 8979 @> WARNING failed to parse beta-factor at line 8980 @> WARNING failed to parse beta-factor at line 8981 @> WARNING failed to parse beta-factor at line 8982 @> WARNING failed to parse beta-factor at line 8983 @> WARNING failed to parse beta-factor at line 8984 @> WARNING failed to parse beta-factor at line 8985 @> WARNING failed to parse beta-factor at line 8986 @> WARNING failed to parse beta-factor at line 8987 @> WARNING failed to parse beta-factor at line 8988 @> WARNING failed to parse beta-factor at line 8989 @> WARNING failed to parse beta-factor at line 8990 @> WARNING failed to parse beta-factor at line 8991 @> WARNING failed to parse beta-factor at line 8992 @> WARNING failed to parse beta-factor at line 8993 @> WARNING failed to parse beta-factor at line 8994 @> WARNING failed to parse beta-factor at line 8995 @> WARNING failed to parse beta-factor at line 8996 @> WARNING failed to parse beta-factor at line 8997 @> WARNING failed to parse beta-factor at line 8998 @> WARNING failed to parse beta-factor at line 8999 @> WARNING failed to parse beta-factor at line 9000 @> WARNING failed to parse beta-factor at line 9001 @> WARNING failed to parse beta-factor at line 9002 @> WARNING failed to parse beta-factor at line 9003 @> WARNING failed to parse beta-factor at line 9004 @> WARNING failed to parse beta-factor at line 9005 @> WARNING failed to parse beta-factor at line 9006 @> WARNING failed to parse beta-factor at line 9007 @> WARNING failed to parse beta-factor at line 9008 @> WARNING failed to parse beta-factor at line 9009 @> WARNING failed to parse beta-factor at line 9010 @> WARNING failed to parse beta-factor at line 9011 @> WARNING failed to parse beta-factor at line 9012 @> WARNING failed to parse beta-factor at line 9013 @> WARNING failed to parse beta-factor at line 9014 @> WARNING failed to parse beta-factor at line 9015 @> WARNING failed to parse beta-factor at line 9016 @> WARNING failed to parse beta-factor at line 9017 @> WARNING failed to parse beta-factor at line 9018 @> WARNING failed to parse beta-factor at line 9019 @> WARNING failed to parse beta-factor at line 9020 @> WARNING failed to parse beta-factor at line 9021 @> WARNING failed to parse beta-factor at line 9022 @> WARNING failed to parse beta-factor at line 9023 @> WARNING failed to parse beta-factor at line 9024 @> WARNING failed to parse beta-factor at line 9025 @> WARNING failed to parse beta-factor at line 9026 @> WARNING failed to parse beta-factor at line 9027 @> WARNING failed to parse beta-factor at line 9028 @> WARNING failed to parse beta-factor at line 9029 @> WARNING failed to parse beta-factor at line 9030 @> WARNING failed to parse beta-factor at line 9031 @> WARNING failed to parse beta-factor at line 9032 @> WARNING failed to parse beta-factor at line 9033 @> WARNING failed to parse beta-factor at line 9034 @> WARNING failed to parse beta-factor at line 9035 @> WARNING failed to parse beta-factor at line 9036 @> WARNING failed to parse beta-factor at line 9037 @> WARNING failed to parse beta-factor at line 9038 @> WARNING failed to parse beta-factor at line 9039 @> WARNING failed to parse beta-factor at line 9040 @> WARNING failed to parse beta-factor at line 9041 @> WARNING failed to parse beta-factor at line 9042 @> WARNING failed to parse beta-factor at line 9043 @> WARNING failed to parse beta-factor at line 9044 @> WARNING failed to parse beta-factor at line 9045 @> WARNING failed to parse beta-factor at line 9046 @> WARNING failed to parse beta-factor at line 9047 @> WARNING failed to parse beta-factor at line 9048 @> WARNING failed to parse beta-factor at line 9049 @> WARNING failed to parse beta-factor at line 9050 @> WARNING failed to parse beta-factor at line 9051 @> WARNING failed to parse beta-factor at line 9052 @> WARNING failed to parse beta-factor at line 9053 @> WARNING failed to parse beta-factor at line 9054 @> WARNING failed to parse beta-factor at line 9055 @> WARNING failed to parse beta-factor at line 9056 @> WARNING failed to parse beta-factor at line 9057 @> WARNING failed to parse beta-factor at line 9058 @> WARNING failed to parse beta-factor at line 9059 @> WARNING failed to parse beta-factor at line 9060 @> WARNING failed to parse beta-factor at line 9061 @> WARNING failed to parse beta-factor at line 9062 @> WARNING failed to parse beta-factor at line 9063 @> WARNING failed to parse beta-factor at line 9064 @> WARNING failed to parse beta-factor at line 9065 @> WARNING failed to parse beta-factor at line 9066 @> WARNING failed to parse beta-factor at line 9067 @> WARNING failed to parse beta-factor at line 9068 @> WARNING failed to parse beta-factor at line 9069 @> WARNING failed to parse beta-factor at line 9070 @> WARNING failed to parse beta-factor at line 9071 @> WARNING failed to parse beta-factor at line 9072 @> WARNING failed to parse beta-factor at line 9073 @> WARNING failed to parse beta-factor at line 9074 @> WARNING failed to parse beta-factor at line 9075 @> WARNING failed to parse beta-factor at line 9076 @> WARNING failed to parse beta-factor at line 9077 @> WARNING failed to parse beta-factor at line 9078 @> WARNING failed to parse beta-factor at line 9079 @> WARNING failed to parse beta-factor at line 9080 @> WARNING failed to parse beta-factor at line 9081 @> WARNING failed to parse beta-factor at line 9082 @> WARNING failed to parse beta-factor at line 9083 @> WARNING failed to parse beta-factor at line 9084 @> WARNING failed to parse beta-factor at line 9085 @> WARNING failed to parse beta-factor at line 9086 @> WARNING failed to parse beta-factor at line 9087 @> WARNING failed to parse beta-factor at line 9088 @> WARNING failed to parse beta-factor at line 9089 @> WARNING failed to parse beta-factor at line 9090 @> WARNING failed to parse beta-factor at line 9091 @> WARNING failed to parse beta-factor at line 9092 @> WARNING failed to parse beta-factor at line 9093 @> WARNING failed to parse beta-factor at line 9094 @> WARNING failed to parse beta-factor at line 9095 @> WARNING failed to parse beta-factor at line 9096 @> WARNING failed to parse beta-factor at line 9097 @> WARNING failed to parse beta-factor at line 9098 @> WARNING failed to parse beta-factor at line 9099 @> WARNING failed to parse beta-factor at line 9100 @> WARNING failed to parse beta-factor at line 9101 @> WARNING failed to parse beta-factor at line 9102 @> WARNING failed to parse beta-factor at line 9103 @> WARNING failed to parse beta-factor at line 9104 @> WARNING failed to parse beta-factor at line 9105 @> WARNING failed to parse beta-factor at line 9106 @> WARNING failed to parse beta-factor at line 9107 @> WARNING failed to parse beta-factor at line 9108 @> WARNING failed to parse beta-factor at line 9109 @> WARNING failed to parse beta-factor at line 9110 @> WARNING failed to parse beta-factor at line 9111 @> WARNING failed to parse beta-factor at line 9112 @> WARNING failed to parse beta-factor at line 9113 @> WARNING failed to parse beta-factor at line 9114 @> WARNING failed to parse beta-factor at line 9115 @> WARNING failed to parse beta-factor at line 9116 @> WARNING failed to parse beta-factor at line 9117 @> WARNING failed to parse beta-factor at line 9118 @> WARNING failed to parse beta-factor at line 9119 @> WARNING failed to parse beta-factor at line 9120 @> WARNING failed to parse beta-factor at line 9121 @> WARNING failed to parse beta-factor at line 9122 @> WARNING failed to parse beta-factor at line 9123 @> WARNING failed to parse beta-factor at line 9124 @> WARNING failed to parse beta-factor at line 9125 @> WARNING failed to parse beta-factor at line 9126 @> WARNING failed to parse beta-factor at line 9127 @> WARNING failed to parse beta-factor at line 9128 @> WARNING failed to parse beta-factor at line 9129 @> WARNING failed to parse beta-factor at line 9130 @> WARNING failed to parse beta-factor at line 9131 @> WARNING failed to parse beta-factor at line 9132 @> WARNING failed to parse beta-factor at line 9133 @> WARNING failed to parse beta-factor at line 9134 @> WARNING failed to parse beta-factor at line 9135 @> WARNING failed to parse beta-factor at line 9136 @> WARNING failed to parse beta-factor at line 9137 @> WARNING failed to parse beta-factor at line 9138 @> WARNING failed to parse beta-factor at line 9139 @> WARNING failed to parse beta-factor at line 9140 @> WARNING failed to parse beta-factor at line 9141 @> WARNING failed to parse beta-factor at line 9142 @> WARNING failed to parse beta-factor at line 9143 @> WARNING failed to parse beta-factor at line 9144 @> WARNING failed to parse beta-factor at line 9145 @> WARNING failed to parse beta-factor at line 9146 @> WARNING failed to parse beta-factor at line 9147 @> WARNING failed to parse beta-factor at line 9148 @> WARNING failed to parse beta-factor at line 9149 @> WARNING failed to parse beta-factor at line 9150 @> WARNING failed to parse beta-factor at line 9151 @> WARNING failed to parse beta-factor at line 9152 @> WARNING failed to parse beta-factor at line 9153 @> WARNING failed to parse beta-factor at line 9154 @> WARNING failed to parse beta-factor at line 9155 @> WARNING failed to parse beta-factor at line 9156 @> WARNING failed to parse beta-factor at line 9157 @> WARNING failed to parse beta-factor at line 9158 @> WARNING failed to parse beta-factor at line 9159 @> WARNING failed to parse beta-factor at line 9160 @> WARNING failed to parse beta-factor at line 9161 @> WARNING failed to parse beta-factor at line 9162 @> WARNING failed to parse beta-factor at line 9163 @> WARNING failed to parse beta-factor at line 9164 @> WARNING failed to parse beta-factor at line 9165 @> WARNING failed to parse beta-factor at line 9166 @> WARNING failed to parse beta-factor at line 9167 @> WARNING failed to parse beta-factor at line 9168 @> WARNING failed to parse beta-factor at line 9169 @> WARNING failed to parse beta-factor at line 9170 @> WARNING failed to parse beta-factor at line 9171 @> WARNING failed to parse beta-factor at line 9172 @> WARNING failed to parse beta-factor at line 9173 @> WARNING failed to parse beta-factor at line 9174 @> WARNING failed to parse beta-factor at line 9175 @> WARNING failed to parse beta-factor at line 9176 @> WARNING failed to parse beta-factor at line 9177 @> WARNING failed to parse beta-factor at line 9178 @> WARNING failed to parse beta-factor at line 9179 @> WARNING failed to parse beta-factor at line 9180 @> WARNING failed to parse beta-factor at line 9181 @> WARNING failed to parse beta-factor at line 9182 @> WARNING failed to parse beta-factor at line 9183 @> WARNING failed to parse beta-factor at line 9184 @> WARNING failed to parse beta-factor at line 9185 @> WARNING failed to parse beta-factor at line 9186 @> WARNING failed to parse beta-factor at line 9187 @> WARNING failed to parse beta-factor at line 9188 @> WARNING failed to parse beta-factor at line 9189 @> WARNING failed to parse beta-factor at line 9190 @> WARNING failed to parse beta-factor at line 9191 @> WARNING failed to parse beta-factor at line 9192 @> WARNING failed to parse beta-factor at line 9193 @> WARNING failed to parse beta-factor at line 9194 @> WARNING failed to parse beta-factor at line 9195 @> WARNING failed to parse beta-factor at line 9196 @> WARNING failed to parse beta-factor at line 9197 @> WARNING failed to parse beta-factor at line 9198 @> WARNING failed to parse beta-factor at line 9199 @> WARNING failed to parse beta-factor at line 9200 @> WARNING failed to parse beta-factor at line 9201 @> WARNING failed to parse beta-factor at line 9202 @> WARNING failed to parse beta-factor at line 9203 @> WARNING failed to parse beta-factor at line 9204 @> WARNING failed to parse beta-factor at line 9205 @> WARNING failed to parse beta-factor at line 9206 @> WARNING failed to parse beta-factor at line 9207 @> WARNING failed to parse beta-factor at line 9208 @> WARNING failed to parse beta-factor at line 9209 @> WARNING failed to parse beta-factor at line 9210 @> WARNING failed to parse beta-factor at line 9211 @> WARNING failed to parse beta-factor at line 9212 @> WARNING failed to parse beta-factor at line 9213 @> WARNING failed to parse beta-factor at line 9214 @> WARNING failed to parse beta-factor at line 9215 @> WARNING failed to parse beta-factor at line 9216 @> WARNING failed to parse beta-factor at line 9217 @> WARNING failed to parse beta-factor at line 9218 @> WARNING failed to parse beta-factor at line 9219 @> WARNING failed to parse beta-factor at line 9220 @> WARNING failed to parse beta-factor at line 9221 @> WARNING failed to parse beta-factor at line 9222 @> WARNING failed to parse beta-factor at line 9223 @> WARNING failed to parse beta-factor at line 9224 @> WARNING failed to parse beta-factor at line 9225 @> WARNING failed to parse beta-factor at line 9226 @> WARNING failed to parse beta-factor at line 9227 @> WARNING failed to parse beta-factor at line 9228 @> WARNING failed to parse beta-factor at line 9229 @> WARNING failed to parse beta-factor at line 9230 @> WARNING failed to parse beta-factor at line 9231 @> WARNING failed to parse beta-factor at line 9232 @> WARNING failed to parse beta-factor at line 9233 @> WARNING failed to parse beta-factor at line 9234 @> WARNING failed to parse beta-factor at line 9235 @> WARNING failed to parse beta-factor at line 9236 @> WARNING failed to parse beta-factor at line 9237 @> WARNING failed to parse beta-factor at line 9238 @> WARNING failed to parse beta-factor at line 9239 @> WARNING failed to parse beta-factor at line 9240 @> WARNING failed to parse beta-factor at line 9241 @> WARNING failed to parse beta-factor at line 9242 @> WARNING failed to parse beta-factor at line 9243 @> WARNING failed to parse beta-factor at line 9244 @> WARNING failed to parse beta-factor at line 9245 @> WARNING failed to parse beta-factor at line 9246 @> WARNING failed to parse beta-factor at line 9247 @> WARNING failed to parse beta-factor at line 9248 @> WARNING failed to parse beta-factor at line 9249 @> WARNING failed to parse beta-factor at line 9250 @> WARNING failed to parse beta-factor at line 9251 @> WARNING failed to parse beta-factor at line 9252 @> WARNING failed to parse beta-factor at line 9253 @> WARNING failed to parse beta-factor at line 9254 @> WARNING failed to parse beta-factor at line 9255 @> WARNING failed to parse beta-factor at line 9256 @> WARNING failed to parse beta-factor at line 9257 @> WARNING failed to parse beta-factor at line 9258 @> WARNING failed to parse beta-factor at line 9259 @> WARNING failed to parse beta-factor at line 9260 @> WARNING failed to parse beta-factor at line 9261 @> WARNING failed to parse beta-factor at line 9262 @> WARNING failed to parse beta-factor at line 9263 @> WARNING failed to parse beta-factor at line 9264 @> WARNING failed to parse beta-factor at line 9265 @> WARNING failed to parse beta-factor at line 9266 @> WARNING failed to parse beta-factor at line 9267 @> WARNING failed to parse beta-factor at line 9268 @> WARNING failed to parse beta-factor at line 9269 @> WARNING failed to parse beta-factor at line 9270 @> WARNING failed to parse beta-factor at line 9271 @> WARNING failed to parse beta-factor at line 9272 @> WARNING failed to parse beta-factor at line 9273 @> WARNING failed to parse beta-factor at line 9274 @> WARNING failed to parse beta-factor at line 9275 @> WARNING failed to parse beta-factor at line 9276 @> WARNING failed to parse beta-factor at line 9277 @> WARNING failed to parse beta-factor at line 9278 @> WARNING failed to parse beta-factor at line 9279 @> WARNING failed to parse beta-factor at line 9280 @> WARNING failed to parse beta-factor at line 9281 @> WARNING failed to parse beta-factor at line 9282 @> WARNING failed to parse beta-factor at line 9283 @> WARNING failed to parse beta-factor at line 9284 @> WARNING failed to parse beta-factor at line 9285 @> WARNING failed to parse beta-factor at line 9286 @> WARNING failed to parse beta-factor at line 9287 @> WARNING failed to parse beta-factor at line 9288 @> WARNING failed to parse beta-factor at line 9289 @> WARNING failed to parse beta-factor at line 9290 @> WARNING failed to parse beta-factor at line 9291 @> WARNING failed to parse beta-factor at line 9292 @> WARNING failed to parse beta-factor at line 9293 @> WARNING failed to parse beta-factor at line 9294 @> WARNING failed to parse beta-factor at line 9295 @> WARNING failed to parse beta-factor at line 9296 @> WARNING failed to parse beta-factor at line 9297 @> WARNING failed to parse beta-factor at line 9298 @> WARNING failed to parse beta-factor at line 9299 @> WARNING failed to parse beta-factor at line 9300 @> WARNING failed to parse beta-factor at line 9301 @> WARNING failed to parse beta-factor at line 9302 @> WARNING failed to parse beta-factor at line 9303 @> WARNING failed to parse beta-factor at line 9304 @> WARNING failed to parse beta-factor at line 9305 @> WARNING failed to parse beta-factor at line 9306 @> WARNING failed to parse beta-factor at line 9307 @> WARNING failed to parse beta-factor at line 9308 @> WARNING failed to parse beta-factor at line 9309 @> WARNING failed to parse beta-factor at line 9310 @> WARNING failed to parse beta-factor at line 9311 @> WARNING failed to parse beta-factor at line 9312 @> WARNING failed to parse beta-factor at line 9313 @> WARNING failed to parse beta-factor at line 9314 @> WARNING failed to parse beta-factor at line 9315 @> WARNING failed to parse beta-factor at line 9316 @> WARNING failed to parse beta-factor at line 9317 @> WARNING failed to parse beta-factor at line 9318 @> WARNING failed to parse beta-factor at line 9319 @> WARNING failed to parse beta-factor at line 9320 @> WARNING failed to parse beta-factor at line 9321 @> WARNING failed to parse beta-factor at line 9322 @> WARNING failed to parse beta-factor at line 9323 @> WARNING failed to parse beta-factor at line 9324 @> WARNING failed to parse beta-factor at line 9325 @> WARNING failed to parse beta-factor at line 9326 @> WARNING failed to parse beta-factor at line 9327 @> WARNING failed to parse beta-factor at line 9328 @> WARNING failed to parse beta-factor at line 9329 @> WARNING failed to parse beta-factor at line 9330 @> WARNING failed to parse beta-factor at line 9331 @> WARNING failed to parse beta-factor at line 9332 @> WARNING failed to parse beta-factor at line 9333 @> WARNING failed to parse beta-factor at line 9334 @> WARNING failed to parse beta-factor at line 9335 @> WARNING failed to parse beta-factor at line 9336 @> WARNING failed to parse beta-factor at line 9337 @> WARNING failed to parse beta-factor at line 9338 @> WARNING failed to parse beta-factor at line 9339 @> WARNING failed to parse beta-factor at line 9340 @> WARNING failed to parse beta-factor at line 9341 @> WARNING failed to parse beta-factor at line 9342 @> WARNING failed to parse beta-factor at line 9343 @> WARNING failed to parse beta-factor at line 9344 @> WARNING failed to parse beta-factor at line 9345 @> WARNING failed to parse beta-factor at line 9346 @> WARNING failed to parse beta-factor at line 9347 @> WARNING failed to parse beta-factor at line 9348 @> WARNING failed to parse beta-factor at line 9349 @> WARNING failed to parse beta-factor at line 9350 @> WARNING failed to parse beta-factor at line 9351 @> WARNING failed to parse beta-factor at line 9352 @> WARNING failed to parse beta-factor at line 9353 @> WARNING failed to parse beta-factor at line 9354 @> WARNING failed to parse beta-factor at line 9355 @> WARNING failed to parse beta-factor at line 9356 @> WARNING failed to parse beta-factor at line 9357 @> WARNING failed to parse beta-factor at line 9358 @> WARNING failed to parse beta-factor at line 9359 @> WARNING failed to parse beta-factor at line 9360 @> WARNING failed to parse beta-factor at line 9361 @> WARNING failed to parse beta-factor at line 9362 @> WARNING failed to parse beta-factor at line 9363 @> WARNING failed to parse beta-factor at line 9364 @> WARNING failed to parse beta-factor at line 9365 @> WARNING failed to parse beta-factor at line 9366 @> WARNING failed to parse beta-factor at line 9367 @> WARNING failed to parse beta-factor at line 9368 @> WARNING failed to parse beta-factor at line 9369 @> WARNING failed to parse beta-factor at line 9370 @> WARNING failed to parse beta-factor at line 9371 @> WARNING failed to parse beta-factor at line 9372 @> WARNING failed to parse beta-factor at line 9373 @> WARNING failed to parse beta-factor at line 9374 @> WARNING failed to parse beta-factor at line 9375 @> WARNING failed to parse beta-factor at line 9376 @> WARNING failed to parse beta-factor at line 9377 @> WARNING failed to parse beta-factor at line 9378 @> WARNING failed to parse beta-factor at line 9379 @> WARNING failed to parse beta-factor at line 9380 @> WARNING failed to parse beta-factor at line 9381 @> WARNING failed to parse beta-factor at line 9382 @> WARNING failed to parse beta-factor at line 9383 @> WARNING failed to parse beta-factor at line 9384 @> WARNING failed to parse beta-factor at line 9385 @> WARNING failed to parse beta-factor at line 9386 @> WARNING failed to parse beta-factor at line 9387 @> WARNING failed to parse beta-factor at line 9388 @> WARNING failed to parse beta-factor at line 9389 @> WARNING failed to parse beta-factor at line 9390 @> WARNING failed to parse beta-factor at line 9391 @> WARNING failed to parse beta-factor at line 9392 @> WARNING failed to parse beta-factor at line 9393 @> WARNING failed to parse beta-factor at line 9394 @> WARNING failed to parse beta-factor at line 9395 @> WARNING failed to parse beta-factor at line 9396 @> WARNING failed to parse beta-factor at line 9397 @> WARNING failed to parse beta-factor at line 9398 @> WARNING failed to parse beta-factor at line 9399 @> WARNING failed to parse beta-factor at line 9400 @> WARNING failed to parse beta-factor at line 9401 @> WARNING failed to parse beta-factor at line 9402 @> WARNING failed to parse beta-factor at line 9403 @> WARNING failed to parse beta-factor at line 9404 @> WARNING failed to parse beta-factor at line 9405 @> WARNING failed to parse beta-factor at line 9406 @> WARNING failed to parse beta-factor at line 9407 @> WARNING failed to parse beta-factor at line 9408 @> WARNING failed to parse beta-factor at line 9409 @> WARNING failed to parse beta-factor at line 9410 @> WARNING failed to parse beta-factor at line 9411 @> WARNING failed to parse beta-factor at line 9412 @> WARNING failed to parse beta-factor at line 9413 @> WARNING failed to parse beta-factor at line 9414 @> WARNING failed to parse beta-factor at line 9415 @> WARNING failed to parse beta-factor at line 9416 @> WARNING failed to parse beta-factor at line 9417 @> WARNING failed to parse beta-factor at line 9418 @> WARNING failed to parse beta-factor at line 9419 @> WARNING failed to parse beta-factor at line 9420 @> WARNING failed to parse beta-factor at line 9421 @> WARNING failed to parse beta-factor at line 9422 @> WARNING failed to parse beta-factor at line 9423 @> WARNING failed to parse beta-factor at line 9424 @> WARNING failed to parse beta-factor at line 9425 @> WARNING failed to parse beta-factor at line 9426 @> WARNING failed to parse beta-factor at line 9427 @> WARNING failed to parse beta-factor at line 9428 @> WARNING failed to parse beta-factor at line 9429 @> WARNING failed to parse beta-factor at line 9430 @> WARNING failed to parse beta-factor at line 9431 @> WARNING failed to parse beta-factor at line 9432 @> WARNING failed to parse beta-factor at line 9433 @> WARNING failed to parse beta-factor at line 9434 @> WARNING failed to parse beta-factor at line 9435 @> WARNING failed to parse beta-factor at line 9436 @> WARNING failed to parse beta-factor at line 9437 @> WARNING failed to parse beta-factor at line 9438 @> WARNING failed to parse beta-factor at line 9439 @> WARNING failed to parse beta-factor at line 9440 @> WARNING failed to parse beta-factor at line 9441 @> WARNING failed to parse beta-factor at line 9442 @> WARNING failed to parse beta-factor at line 9443 @> WARNING failed to parse beta-factor at line 9444 @> WARNING failed to parse beta-factor at line 9445 @> WARNING failed to parse beta-factor at line 9446 @> WARNING failed to parse beta-factor at line 9447 @> WARNING failed to parse beta-factor at line 9448 @> WARNING failed to parse beta-factor at line 9449 @> WARNING failed to parse beta-factor at line 9450 @> WARNING failed to parse beta-factor at line 9451 @> WARNING failed to parse beta-factor at line 9452 @> WARNING failed to parse beta-factor at line 9453 @> WARNING failed to parse beta-factor at line 9454 @> WARNING failed to parse beta-factor at line 9455 @> WARNING failed to parse beta-factor at line 9456 @> WARNING failed to parse beta-factor at line 9457 @> WARNING failed to parse beta-factor at line 9458 @> WARNING failed to parse beta-factor at line 9459 @> WARNING failed to parse beta-factor at line 9460 @> WARNING failed to parse beta-factor at line 9461 @> WARNING failed to parse beta-factor at line 9462 @> WARNING failed to parse beta-factor at line 9463 @> WARNING failed to parse beta-factor at line 9464 @> WARNING failed to parse beta-factor at line 9465 @> WARNING failed to parse beta-factor at line 9466 @> WARNING failed to parse beta-factor at line 9467 @> WARNING failed to parse beta-factor at line 9468 @> WARNING failed to parse beta-factor at line 9469 @> WARNING failed to parse beta-factor at line 9470 @> WARNING failed to parse beta-factor at line 9471 @> WARNING failed to parse beta-factor at line 9472 @> WARNING failed to parse beta-factor at line 9473 @> WARNING failed to parse beta-factor at line 9474 @> WARNING failed to parse beta-factor at line 9475 @> WARNING failed to parse beta-factor at line 9476 @> WARNING failed to parse beta-factor at line 9477 @> WARNING failed to parse beta-factor at line 9478 @> WARNING failed to parse beta-factor at line 9479 @> WARNING failed to parse beta-factor at line 9480 @> WARNING failed to parse beta-factor at line 9481 @> WARNING failed to parse beta-factor at line 9482 @> WARNING failed to parse beta-factor at line 9483 @> WARNING failed to parse beta-factor at line 9484 @> WARNING failed to parse beta-factor at line 9485 @> WARNING failed to parse beta-factor at line 9486 @> WARNING failed to parse beta-factor at line 9487 @> WARNING failed to parse beta-factor at line 9488 @> WARNING failed to parse beta-factor at line 9489 @> WARNING failed to parse beta-factor at line 9490 @> WARNING failed to parse beta-factor at line 9491 @> WARNING failed to parse beta-factor at line 9492 @> WARNING failed to parse beta-factor at line 9493 @> WARNING failed to parse beta-factor at line 9494 @> WARNING failed to parse beta-factor at line 9495 @> WARNING failed to parse beta-factor at line 9496 @> WARNING failed to parse beta-factor at line 9497 @> WARNING failed to parse beta-factor at line 9498 @> WARNING failed to parse beta-factor at line 9499 @> WARNING failed to parse beta-factor at line 9500 @> WARNING failed to parse beta-factor at line 9501 @> WARNING failed to parse beta-factor at line 9502 @> WARNING failed to parse beta-factor at line 9503 @> WARNING failed to parse beta-factor at line 9504 @> WARNING failed to parse beta-factor at line 9505 @> WARNING failed to parse beta-factor at line 9506 @> WARNING failed to parse beta-factor at line 9507 @> WARNING failed to parse beta-factor at line 9508 @> WARNING failed to parse beta-factor at line 9509 @> WARNING failed to parse beta-factor at line 9510 @> WARNING failed to parse beta-factor at line 9511 @> WARNING failed to parse beta-factor at line 9512 @> WARNING failed to parse beta-factor at line 9513 @> WARNING failed to parse beta-factor at line 9514 @> WARNING failed to parse beta-factor at line 9515 @> WARNING failed to parse beta-factor at line 9516 @> WARNING failed to parse beta-factor at line 9517 @> WARNING failed to parse beta-factor at line 9518 @> WARNING failed to parse beta-factor at line 9519 @> WARNING failed to parse beta-factor at line 9520 @> WARNING failed to parse beta-factor at line 9521 @> WARNING failed to parse beta-factor at line 9522 @> WARNING failed to parse beta-factor at line 9523 @> WARNING failed to parse beta-factor at line 9524 @> WARNING failed to parse beta-factor at line 9525 @> WARNING failed to parse beta-factor at line 9526 @> WARNING failed to parse beta-factor at line 9527 @> WARNING failed to parse beta-factor at line 9528 @> WARNING failed to parse beta-factor at line 9529 @> WARNING failed to parse beta-factor at line 9530 @> WARNING failed to parse beta-factor at line 9531 @> WARNING failed to parse beta-factor at line 9532 @> WARNING failed to parse beta-factor at line 9533 @> WARNING failed to parse beta-factor at line 9534 @> WARNING failed to parse beta-factor at line 9535 @> WARNING failed to parse beta-factor at line 9536 @> WARNING failed to parse beta-factor at line 9537 @> WARNING failed to parse beta-factor at line 9538 @> WARNING failed to parse beta-factor at line 9539 @> WARNING failed to parse beta-factor at line 9540 @> WARNING failed to parse beta-factor at line 9541 @> WARNING failed to parse beta-factor at line 9542 @> WARNING failed to parse beta-factor at line 9543 @> WARNING failed to parse beta-factor at line 9544 @> WARNING failed to parse beta-factor at line 9545 @> WARNING failed to parse beta-factor at line 9546 @> WARNING failed to parse beta-factor at line 9547 @> WARNING failed to parse beta-factor at line 9548 @> WARNING failed to parse beta-factor at line 9549 @> WARNING failed to parse beta-factor at line 9550 @> WARNING failed to parse beta-factor at line 9551 @> WARNING failed to parse beta-factor at line 9552 @> WARNING failed to parse beta-factor at line 9553 @> WARNING failed to parse beta-factor at line 9554 @> WARNING failed to parse beta-factor at line 9555 @> WARNING failed to parse beta-factor at line 9556 @> WARNING failed to parse beta-factor at line 9557 @> WARNING failed to parse beta-factor at line 9558 @> WARNING failed to parse beta-factor at line 9559 @> WARNING failed to parse beta-factor at line 9560 @> WARNING failed to parse beta-factor at line 9561 @> WARNING failed to parse beta-factor at line 9562 @> WARNING failed to parse beta-factor at line 9563 @> WARNING failed to parse beta-factor at line 9564 @> WARNING failed to parse beta-factor at line 9565 @> WARNING failed to parse beta-factor at line 9566 @> WARNING failed to parse beta-factor at line 9567 @> WARNING failed to parse beta-factor at line 9568 @> WARNING failed to parse beta-factor at line 9569 @> WARNING failed to parse beta-factor at line 9570 @> WARNING failed to parse beta-factor at line 9571 @> WARNING failed to parse beta-factor at line 9572 @> WARNING failed to parse beta-factor at line 9573 @> WARNING failed to parse beta-factor at line 9574 @> WARNING failed to parse beta-factor at line 9575 @> WARNING failed to parse beta-factor at line 9576 @> WARNING failed to parse beta-factor at line 9577 @> WARNING failed to parse beta-factor at line 9578 @> WARNING failed to parse beta-factor at line 9579 @> WARNING failed to parse beta-factor at line 9580 @> WARNING failed to parse beta-factor at line 9581 @> WARNING failed to parse beta-factor at line 9582 @> WARNING failed to parse beta-factor at line 9583 @> WARNING failed to parse beta-factor at line 9584 @> WARNING failed to parse beta-factor at line 9585 @> WARNING failed to parse beta-factor at line 9586 @> WARNING failed to parse beta-factor at line 9587 @> WARNING failed to parse beta-factor at line 9588 @> WARNING failed to parse beta-factor at line 9589 @> WARNING failed to parse beta-factor at line 9590 @> WARNING failed to parse beta-factor at line 9591 @> WARNING failed to parse beta-factor at line 9592 @> WARNING failed to parse beta-factor at line 9593 @> WARNING failed to parse beta-factor at line 9594 @> WARNING failed to parse beta-factor at line 9595 @> WARNING failed to parse beta-factor at line 9596 @> WARNING failed to parse beta-factor at line 9597 @> WARNING failed to parse beta-factor at line 9598 @> WARNING failed to parse beta-factor at line 9599 @> WARNING failed to parse beta-factor at line 9600 @> WARNING failed to parse beta-factor at line 9601 @> WARNING failed to parse beta-factor at line 9602 @> WARNING failed to parse beta-factor at line 9603 @> WARNING failed to parse beta-factor at line 9604 @> WARNING failed to parse beta-factor at line 9605 @> WARNING failed to parse beta-factor at line 9606 @> WARNING failed to parse beta-factor at line 9607 @> WARNING failed to parse beta-factor at line 9608 @> WARNING failed to parse beta-factor at line 9609 @> WARNING failed to parse beta-factor at line 9610 @> WARNING failed to parse beta-factor at line 9611 @> WARNING failed to parse beta-factor at line 9612 @> WARNING failed to parse beta-factor at line 9613 @> WARNING failed to parse beta-factor at line 9614 @> WARNING failed to parse beta-factor at line 9615 @> WARNING failed to parse beta-factor at line 9616 @> WARNING failed to parse beta-factor at line 9617 @> WARNING failed to parse beta-factor at line 9618 @> WARNING failed to parse beta-factor at line 9619 @> WARNING failed to parse beta-factor at line 9620 @> WARNING failed to parse beta-factor at line 9621 @> WARNING failed to parse beta-factor at line 9622 @> WARNING failed to parse beta-factor at line 9623 @> WARNING failed to parse beta-factor at line 9624 @> WARNING failed to parse beta-factor at line 9625 @> WARNING failed to parse beta-factor at line 9626 @> WARNING failed to parse beta-factor at line 9627 @> WARNING failed to parse beta-factor at line 9628 @> WARNING failed to parse beta-factor at line 9629 @> WARNING failed to parse beta-factor at line 9630 @> WARNING failed to parse beta-factor at line 9631 @> WARNING failed to parse beta-factor at line 9632 @> WARNING failed to parse beta-factor at line 9633 @> WARNING failed to parse beta-factor at line 9634 @> WARNING failed to parse beta-factor at line 9635 @> WARNING failed to parse beta-factor at line 9636 @> WARNING failed to parse beta-factor at line 9637 @> WARNING failed to parse beta-factor at line 9638 @> WARNING failed to parse beta-factor at line 9639 @> WARNING failed to parse beta-factor at line 9640 @> WARNING failed to parse beta-factor at line 9641 @> WARNING failed to parse beta-factor at line 9642 @> WARNING failed to parse beta-factor at line 9643 @> WARNING failed to parse beta-factor at line 9644 @> WARNING failed to parse beta-factor at line 9645 @> WARNING failed to parse beta-factor at line 9646 @> WARNING failed to parse beta-factor at line 9647 @> WARNING failed to parse beta-factor at line 9648 @> WARNING failed to parse beta-factor at line 9649 @> WARNING failed to parse beta-factor at line 9650 @> WARNING failed to parse beta-factor at line 9651 @> WARNING failed to parse beta-factor at line 9652 @> WARNING failed to parse beta-factor at line 9653 @> WARNING failed to parse beta-factor at line 9654 @> WARNING failed to parse beta-factor at line 9655 @> WARNING failed to parse beta-factor at line 9656 @> WARNING failed to parse beta-factor at line 9657 @> WARNING failed to parse beta-factor at line 9658 @> WARNING failed to parse beta-factor at line 9659 @> WARNING failed to parse beta-factor at line 9660 @> WARNING failed to parse beta-factor at line 9661 @> WARNING failed to parse beta-factor at line 9662 @> WARNING failed to parse beta-factor at line 9663 @> WARNING failed to parse beta-factor at line 9664 @> WARNING failed to parse beta-factor at line 9665 @> WARNING failed to parse beta-factor at line 9666 @> WARNING failed to parse beta-factor at line 9667 @> WARNING failed to parse beta-factor at line 9668 @> WARNING failed to parse beta-factor at line 9669 @> WARNING failed to parse beta-factor at line 9670 @> WARNING failed to parse beta-factor at line 9671 @> WARNING failed to parse beta-factor at line 9672 @> WARNING failed to parse beta-factor at line 9673 @> WARNING failed to parse beta-factor at line 9674 @> WARNING failed to parse beta-factor at line 9675 @> WARNING failed to parse beta-factor at line 9676 @> WARNING failed to parse beta-factor at line 9677 @> WARNING failed to parse beta-factor at line 9678 @> WARNING failed to parse beta-factor at line 9679 @> WARNING failed to parse beta-factor at line 9680 @> WARNING failed to parse beta-factor at line 9681 @> WARNING failed to parse beta-factor at line 9682 @> WARNING failed to parse beta-factor at line 9683 @> WARNING failed to parse beta-factor at line 9684 @> WARNING failed to parse beta-factor at line 9685 @> WARNING failed to parse beta-factor at line 9686 @> WARNING failed to parse beta-factor at line 9687 @> WARNING failed to parse beta-factor at line 9688 @> WARNING failed to parse beta-factor at line 9689 @> WARNING failed to parse beta-factor at line 9690 @> WARNING failed to parse beta-factor at line 9691 @> WARNING failed to parse beta-factor at line 9692 @> WARNING failed to parse beta-factor at line 9693 @> WARNING failed to parse beta-factor at line 9694 @> WARNING failed to parse beta-factor at line 9695 @> WARNING failed to parse beta-factor at line 9696 @> WARNING failed to parse beta-factor at line 9697 @> WARNING failed to parse beta-factor at line 9698 @> WARNING failed to parse beta-factor at line 9699 @> WARNING failed to parse beta-factor at line 9700 @> WARNING failed to parse beta-factor at line 9701 @> WARNING failed to parse beta-factor at line 9702 @> WARNING failed to parse beta-factor at line 9703 @> WARNING failed to parse beta-factor at line 9704 @> WARNING failed to parse beta-factor at line 9705 @> WARNING failed to parse beta-factor at line 9706 @> WARNING failed to parse beta-factor at line 9707 @> WARNING failed to parse beta-factor at line 9708 @> WARNING failed to parse beta-factor at line 9709 @> WARNING failed to parse beta-factor at line 9710 @> WARNING failed to parse beta-factor at line 9711 @> WARNING failed to parse beta-factor at line 9712 @> WARNING failed to parse beta-factor at line 9713 @> WARNING failed to parse beta-factor at line 9714 @> WARNING failed to parse beta-factor at line 9715 @> WARNING failed to parse beta-factor at line 9716 @> WARNING failed to parse beta-factor at line 9717 @> WARNING failed to parse beta-factor at line 9718 @> WARNING failed to parse beta-factor at line 9719 @> WARNING failed to parse beta-factor at line 9720 @> WARNING failed to parse beta-factor at line 9721 @> WARNING failed to parse beta-factor at line 9722 @> WARNING failed to parse beta-factor at line 9723 @> WARNING failed to parse beta-factor at line 9724 @> WARNING failed to parse beta-factor at line 9725 @> WARNING failed to parse beta-factor at line 9726 @> WARNING failed to parse beta-factor at line 9727 @> WARNING failed to parse beta-factor at line 9728 @> WARNING failed to parse beta-factor at line 9729 @> WARNING failed to parse beta-factor at line 9730 @> WARNING failed to parse beta-factor at line 9731 @> WARNING failed to parse beta-factor at line 9732 @> WARNING failed to parse beta-factor at line 9733 @> WARNING failed to parse beta-factor at line 9734 @> WARNING failed to parse beta-factor at line 9735 @> WARNING failed to parse beta-factor at line 9736 @> WARNING failed to parse beta-factor at line 9737 @> WARNING failed to parse beta-factor at line 9738 @> WARNING failed to parse beta-factor at line 9739 @> WARNING failed to parse beta-factor at line 9740 @> WARNING failed to parse beta-factor at line 9741 @> WARNING failed to parse beta-factor at line 9742 @> WARNING failed to parse beta-factor at line 9743 @> WARNING failed to parse beta-factor at line 9744 @> WARNING failed to parse beta-factor at line 9745 @> WARNING failed to parse beta-factor at line 9746 @> WARNING failed to parse beta-factor at line 9747 @> WARNING failed to parse beta-factor at line 9748 @> WARNING failed to parse beta-factor at line 9749 @> WARNING failed to parse beta-factor at line 9750 @> WARNING failed to parse beta-factor at line 9751 @> WARNING failed to parse beta-factor at line 9752 @> WARNING failed to parse beta-factor at line 9753 @> WARNING failed to parse beta-factor at line 9754 @> WARNING failed to parse beta-factor at line 9755 @> WARNING failed to parse beta-factor at line 9756 @> WARNING failed to parse beta-factor at line 9757 @> WARNING failed to parse beta-factor at line 9758 @> WARNING failed to parse beta-factor at line 9759 @> WARNING failed to parse beta-factor at line 9760 @> WARNING failed to parse beta-factor at line 9761 @> WARNING failed to parse beta-factor at line 9762 @> WARNING failed to parse beta-factor at line 9763 @> WARNING failed to parse beta-factor at line 9764 @> WARNING failed to parse beta-factor at line 9765 @> WARNING failed to parse beta-factor at line 9766 @> WARNING failed to parse beta-factor at line 9767 @> WARNING failed to parse beta-factor at line 9768 @> WARNING failed to parse beta-factor at line 9769 @> WARNING failed to parse beta-factor at line 9770 @> WARNING failed to parse beta-factor at line 9771 @> WARNING failed to parse beta-factor at line 9772 @> WARNING failed to parse beta-factor at line 9773 @> WARNING failed to parse beta-factor at line 9774 @> WARNING failed to parse beta-factor at line 9775 @> WARNING failed to parse beta-factor at line 9776 @> WARNING failed to parse beta-factor at line 9777 @> WARNING failed to parse beta-factor at line 9778 @> WARNING failed to parse beta-factor at line 9779 @> WARNING failed to parse beta-factor at line 9780 @> WARNING failed to parse beta-factor at line 9781 @> WARNING failed to parse beta-factor at line 9782 @> WARNING failed to parse beta-factor at line 9783 @> WARNING failed to parse beta-factor at line 9784 @> WARNING failed to parse beta-factor at line 9785 @> WARNING failed to parse beta-factor at line 9786 @> WARNING failed to parse beta-factor at line 9787 @> WARNING failed to parse beta-factor at line 9788 @> WARNING failed to parse beta-factor at line 9789 @> WARNING failed to parse beta-factor at line 9790 @> WARNING failed to parse beta-factor at line 9791 @> WARNING failed to parse beta-factor at line 9792 @> WARNING failed to parse beta-factor at line 9793 @> WARNING failed to parse beta-factor at line 9794 @> WARNING failed to parse beta-factor at line 9795 @> WARNING failed to parse beta-factor at line 9796 @> WARNING failed to parse beta-factor at line 9797 @> WARNING failed to parse beta-factor at line 9798 @> WARNING failed to parse beta-factor at line 9799 @> WARNING failed to parse beta-factor at line 9800 @> WARNING failed to parse beta-factor at line 9801 @> WARNING failed to parse beta-factor at line 9802 @> WARNING failed to parse beta-factor at line 9803 @> WARNING failed to parse beta-factor at line 9804 @> WARNING failed to parse beta-factor at line 9805 @> WARNING failed to parse beta-factor at line 9806 @> WARNING failed to parse beta-factor at line 9807 @> WARNING failed to parse beta-factor at line 9808 @> WARNING failed to parse beta-factor at line 9809 @> WARNING failed to parse beta-factor at line 9810 @> WARNING failed to parse beta-factor at line 9811 @> WARNING failed to parse beta-factor at line 9812 @> WARNING failed to parse beta-factor at line 9813 @> WARNING failed to parse beta-factor at line 9814 @> WARNING failed to parse beta-factor at line 9815 @> WARNING failed to parse beta-factor at line 9816 @> WARNING failed to parse beta-factor at line 9817 @> WARNING failed to parse beta-factor at line 9818 @> WARNING failed to parse beta-factor at line 9819 @> WARNING failed to parse beta-factor at line 9820 @> WARNING failed to parse beta-factor at line 9821 @> WARNING failed to parse beta-factor at line 9822 @> WARNING failed to parse beta-factor at line 9823 @> WARNING failed to parse beta-factor at line 9824 @> WARNING failed to parse beta-factor at line 9825 @> WARNING failed to parse beta-factor at line 9826 @> WARNING failed to parse beta-factor at line 9827 @> WARNING failed to parse beta-factor at line 9828 @> WARNING failed to parse beta-factor at line 9829 @> WARNING failed to parse beta-factor at line 9830 @> WARNING failed to parse beta-factor at line 9831 @> WARNING failed to parse beta-factor at line 9832 @> WARNING failed to parse beta-factor at line 9833 @> WARNING failed to parse beta-factor at line 9834 @> WARNING failed to parse beta-factor at line 9835 @> WARNING failed to parse beta-factor at line 9836 @> WARNING failed to parse beta-factor at line 9837 @> WARNING failed to parse beta-factor at line 9838 @> WARNING failed to parse beta-factor at line 9839 @> WARNING failed to parse beta-factor at line 9840 @> WARNING failed to parse beta-factor at line 9841 @> WARNING failed to parse beta-factor at line 9842 @> WARNING failed to parse beta-factor at line 9843 @> WARNING failed to parse beta-factor at line 9844 @> WARNING failed to parse beta-factor at line 9845 @> WARNING failed to parse beta-factor at line 9846 @> WARNING failed to parse beta-factor at line 9847 @> WARNING failed to parse beta-factor at line 9848 @> WARNING failed to parse beta-factor at line 9849 @> WARNING failed to parse beta-factor at line 9850 @> WARNING failed to parse beta-factor at line 9851 @> WARNING failed to parse beta-factor at line 9852 @> WARNING failed to parse beta-factor at line 9853 @> WARNING failed to parse beta-factor at line 9854 @> WARNING failed to parse beta-factor at line 9855 @> WARNING failed to parse beta-factor at line 9856 @> WARNING failed to parse beta-factor at line 9857 @> WARNING failed to parse beta-factor at line 9858 @> WARNING failed to parse beta-factor at line 9859 @> WARNING failed to parse beta-factor at line 9860 @> WARNING failed to parse beta-factor at line 9861 @> WARNING failed to parse beta-factor at line 9862 @> WARNING failed to parse beta-factor at line 9863 @> WARNING failed to parse beta-factor at line 9864 @> WARNING failed to parse beta-factor at line 9865 @> WARNING failed to parse beta-factor at line 9866 @> WARNING failed to parse beta-factor at line 9867 @> WARNING failed to parse beta-factor at line 9868 @> WARNING failed to parse beta-factor at line 9869 @> WARNING failed to parse beta-factor at line 9870 @> WARNING failed to parse beta-factor at line 9871 @> WARNING failed to parse beta-factor at line 9872 @> WARNING failed to parse beta-factor at line 9873 @> WARNING failed to parse beta-factor at line 9874 @> WARNING failed to parse beta-factor at line 9875 @> WARNING failed to parse beta-factor at line 9876 @> WARNING failed to parse beta-factor at line 9877 @> WARNING failed to parse beta-factor at line 9878 @> WARNING failed to parse beta-factor at line 9879 @> WARNING failed to parse beta-factor at line 9880 @> WARNING failed to parse beta-factor at line 9881 @> WARNING failed to parse beta-factor at line 9882 @> WARNING failed to parse beta-factor at line 9883 @> WARNING failed to parse beta-factor at line 9884 @> WARNING failed to parse beta-factor at line 9885 @> WARNING failed to parse beta-factor at line 9886 @> WARNING failed to parse beta-factor at line 9887 @> WARNING failed to parse beta-factor at line 9888 @> WARNING failed to parse beta-factor at line 9889 @> WARNING failed to parse beta-factor at line 9890 @> WARNING failed to parse beta-factor at line 9891 @> WARNING failed to parse beta-factor at line 9892 @> WARNING failed to parse beta-factor at line 9893 @> WARNING failed to parse beta-factor at line 9894 @> WARNING failed to parse beta-factor at line 9895 @> WARNING failed to parse beta-factor at line 9896 @> WARNING failed to parse beta-factor at line 9897 @> WARNING failed to parse beta-factor at line 9898 @> WARNING failed to parse beta-factor at line 9899 @> WARNING failed to parse beta-factor at line 9900 @> WARNING failed to parse beta-factor at line 9901 @> WARNING failed to parse beta-factor at line 9902 @> WARNING failed to parse beta-factor at line 9903 @> WARNING failed to parse beta-factor at line 9904 @> WARNING failed to parse beta-factor at line 9905 @> WARNING failed to parse beta-factor at line 9906 @> WARNING failed to parse beta-factor at line 9907 @> WARNING failed to parse beta-factor at line 9908 @> WARNING failed to parse beta-factor at line 9909 @> WARNING failed to parse beta-factor at line 9910 @> WARNING failed to parse beta-factor at line 9911 @> WARNING failed to parse beta-factor at line 9912 @> WARNING failed to parse beta-factor at line 9913 @> WARNING failed to parse beta-factor at line 9914 @> WARNING failed to parse beta-factor at line 9915 @> WARNING failed to parse beta-factor at line 9916 @> WARNING failed to parse beta-factor at line 9917 @> WARNING failed to parse beta-factor at line 9918 @> WARNING failed to parse beta-factor at line 9919 @> WARNING failed to parse beta-factor at line 9920 @> WARNING failed to parse beta-factor at line 9921 @> WARNING failed to parse beta-factor at line 9922 @> WARNING failed to parse beta-factor at line 9923 @> WARNING failed to parse beta-factor at line 9924 @> WARNING failed to parse beta-factor at line 9925 @> WARNING failed to parse beta-factor at line 9926 @> WARNING failed to parse beta-factor at line 9927 @> WARNING failed to parse beta-factor at line 9928 @> WARNING failed to parse beta-factor at line 9929 @> WARNING failed to parse beta-factor at line 9930 @> WARNING failed to parse beta-factor at line 9931 @> WARNING failed to parse beta-factor at line 9932 @> WARNING failed to parse beta-factor at line 9933 @> WARNING failed to parse beta-factor at line 9934 @> WARNING failed to parse beta-factor at line 9935 @> WARNING failed to parse beta-factor at line 9936 @> WARNING failed to parse beta-factor at line 9937 @> WARNING failed to parse beta-factor at line 9938 @> WARNING failed to parse beta-factor at line 9939 @> WARNING failed to parse beta-factor at line 9940 @> WARNING failed to parse beta-factor at line 9941 @> WARNING failed to parse beta-factor at line 9942 @> WARNING failed to parse beta-factor at line 9943 @> WARNING failed to parse beta-factor at line 9944 @> WARNING failed to parse beta-factor at line 9945 @> WARNING failed to parse beta-factor at line 9946 @> WARNING failed to parse beta-factor at line 9947 @> WARNING failed to parse beta-factor at line 9948 @> WARNING failed to parse beta-factor at line 9949 @> WARNING failed to parse beta-factor at line 9950 @> WARNING failed to parse beta-factor at line 9951 @> WARNING failed to parse beta-factor at line 9952 @> WARNING failed to parse beta-factor at line 9953 @> WARNING failed to parse beta-factor at line 9954 @> WARNING failed to parse beta-factor at line 9955 @> WARNING failed to parse beta-factor at line 9956 @> WARNING failed to parse beta-factor at line 9957 @> WARNING failed to parse beta-factor at line 9958 @> WARNING failed to parse beta-factor at line 9959 @> WARNING failed to parse beta-factor at line 9960 @> WARNING failed to parse beta-factor at line 9961 @> WARNING failed to parse beta-factor at line 9962 @> WARNING failed to parse beta-factor at line 9963 @> WARNING failed to parse beta-factor at line 9964 @> WARNING failed to parse beta-factor at line 9965 @> WARNING failed to parse beta-factor at line 9966 @> WARNING failed to parse beta-factor at line 9967 @> WARNING failed to parse beta-factor at line 9968 @> WARNING failed to parse beta-factor at line 9969 @> WARNING failed to parse beta-factor at line 9970 @> WARNING failed to parse beta-factor at line 9971 @> WARNING failed to parse beta-factor at line 9972 @> WARNING failed to parse beta-factor at line 9973 @> WARNING failed to parse beta-factor at line 9974 @> WARNING failed to parse beta-factor at line 9975 @> WARNING failed to parse beta-factor at line 9976 @> WARNING failed to parse beta-factor at line 9977 @> WARNING failed to parse beta-factor at line 9978 @> WARNING failed to parse beta-factor at line 9979 @> WARNING failed to parse beta-factor at line 9980 @> WARNING failed to parse beta-factor at line 9981 @> WARNING failed to parse beta-factor at line 9982 @> WARNING failed to parse beta-factor at line 9983 @> WARNING failed to parse beta-factor at line 9984 @> WARNING failed to parse beta-factor at line 9985 @> WARNING failed to parse beta-factor at line 9986 @> WARNING failed to parse beta-factor at line 9987 @> WARNING failed to parse beta-factor at line 9988 @> WARNING failed to parse beta-factor at line 9989 @> WARNING failed to parse beta-factor at line 9990 @> WARNING failed to parse beta-factor at line 9991 @> WARNING failed to parse beta-factor at line 9992 @> WARNING failed to parse beta-factor at line 9993 @> WARNING failed to parse beta-factor at line 9994 @> WARNING failed to parse beta-factor at line 9995 @> WARNING failed to parse beta-factor at line 9996 @> WARNING failed to parse beta-factor at line 9997 @> WARNING failed to parse beta-factor at line 9998 @> WARNING failed to parse beta-factor at line 9999 @> WARNING failed to parse beta-factor at line 10000 @> WARNING failed to parse beta-factor at line 10001 @> WARNING failed to parse beta-factor at line 10002 @> WARNING failed to parse beta-factor at line 10003 @> WARNING failed to parse beta-factor at line 10004 @> WARNING failed to parse beta-factor at line 10005 @> WARNING failed to parse beta-factor at line 10006 @> WARNING failed to parse beta-factor at line 10007 @> WARNING failed to parse beta-factor at line 10008 @> WARNING failed to parse beta-factor at line 10009 @> WARNING failed to parse beta-factor at line 10010 @> WARNING failed to parse beta-factor at line 10011 @> WARNING failed to parse beta-factor at line 10012 @> WARNING failed to parse beta-factor at line 10013 @> WARNING failed to parse beta-factor at line 10014 @> WARNING failed to parse beta-factor at line 10015 @> WARNING failed to parse beta-factor at line 10016 @> WARNING failed to parse beta-factor at line 10017 @> WARNING failed to parse beta-factor at line 10018 @> WARNING failed to parse beta-factor at line 10019 @> WARNING failed to parse beta-factor at line 10020 @> WARNING failed to parse beta-factor at line 10021 @> WARNING failed to parse beta-factor at line 10022 @> WARNING failed to parse beta-factor at line 10023 @> WARNING failed to parse beta-factor at line 10024 @> WARNING failed to parse beta-factor at line 10025 @> WARNING failed to parse beta-factor at line 10026 @> WARNING failed to parse beta-factor at line 10027 @> WARNING failed to parse beta-factor at line 10028 @> WARNING failed to parse beta-factor at line 10029 @> WARNING failed to parse beta-factor at line 10030 @> WARNING failed to parse beta-factor at line 10031 @> WARNING failed to parse beta-factor at line 10032 @> WARNING failed to parse beta-factor at line 10033 @> WARNING failed to parse beta-factor at line 10034 @> WARNING failed to parse beta-factor at line 10035 @> WARNING failed to parse beta-factor at line 10036 @> WARNING failed to parse beta-factor at line 10037 @> WARNING failed to parse beta-factor at line 10038 @> WARNING failed to parse beta-factor at line 10039 @> WARNING failed to parse beta-factor at line 10040 @> WARNING failed to parse beta-factor at line 10041 @> WARNING failed to parse beta-factor at line 10042 @> WARNING failed to parse beta-factor at line 10043 @> WARNING failed to parse beta-factor at line 10044 @> WARNING failed to parse beta-factor at line 10045 @> WARNING failed to parse beta-factor at line 10046 @> WARNING failed to parse beta-factor at line 10047 @> WARNING failed to parse beta-factor at line 10048 @> WARNING failed to parse beta-factor at line 10049 @> WARNING failed to parse beta-factor at line 10050 @> WARNING failed to parse beta-factor at line 10051 @> WARNING failed to parse beta-factor at line 10052 @> WARNING failed to parse beta-factor at line 10053 @> WARNING failed to parse beta-factor at line 10054 @> WARNING failed to parse beta-factor at line 10055 @> WARNING failed to parse beta-factor at line 10056 @> WARNING failed to parse beta-factor at line 10057 @> WARNING failed to parse beta-factor at line 10058 @> WARNING failed to parse beta-factor at line 10059 @> WARNING failed to parse beta-factor at line 10060 @> WARNING failed to parse beta-factor at line 10061 @> WARNING failed to parse beta-factor at line 10062 @> WARNING failed to parse beta-factor at line 10063 @> WARNING failed to parse beta-factor at line 10064 @> WARNING failed to parse beta-factor at line 10065 @> WARNING failed to parse beta-factor at line 10066 @> WARNING failed to parse beta-factor at line 10067 @> WARNING failed to parse beta-factor at line 10068 @> WARNING failed to parse beta-factor at line 10069 @> WARNING failed to parse beta-factor at line 10070 @> WARNING failed to parse beta-factor at line 10071 @> WARNING failed to parse beta-factor at line 10072 @> WARNING failed to parse beta-factor at line 10073 @> WARNING failed to parse beta-factor at line 10074 @> WARNING failed to parse beta-factor at line 10075 @> WARNING failed to parse beta-factor at line 10076 @> WARNING failed to parse beta-factor at line 10077 @> WARNING failed to parse beta-factor at line 10078 @> WARNING failed to parse beta-factor at line 10079 @> WARNING failed to parse beta-factor at line 10080 @> WARNING failed to parse beta-factor at line 10081 @> WARNING failed to parse beta-factor at line 10082 @> WARNING failed to parse beta-factor at line 10083 @> WARNING failed to parse beta-factor at line 10084 @> WARNING failed to parse beta-factor at line 10085 @> WARNING failed to parse beta-factor at line 10086 @> WARNING failed to parse beta-factor at line 10087 @> WARNING failed to parse beta-factor at line 10088 @> WARNING failed to parse beta-factor at line 10089 @> WARNING failed to parse beta-factor at line 10090 @> WARNING failed to parse beta-factor at line 10091 @> WARNING failed to parse beta-factor at line 10092 @> WARNING failed to parse beta-factor at line 10093 @> WARNING failed to parse beta-factor at line 10094 @> WARNING failed to parse beta-factor at line 10095 @> WARNING failed to parse beta-factor at line 10096 @> WARNING failed to parse beta-factor at line 10097 @> WARNING failed to parse beta-factor at line 10098 @> WARNING failed to parse beta-factor at line 10099 @> WARNING failed to parse beta-factor at line 10100 @> WARNING failed to parse beta-factor at line 10101 @> WARNING failed to parse beta-factor at line 10102 @> WARNING failed to parse beta-factor at line 10103 @> WARNING failed to parse beta-factor at line 10104 @> WARNING failed to parse beta-factor at line 10105 @> WARNING failed to parse beta-factor at line 10106 @> WARNING failed to parse beta-factor at line 10107 @> WARNING failed to parse beta-factor at line 10108 @> WARNING failed to parse beta-factor at line 10109 @> WARNING failed to parse beta-factor at line 10110 @> WARNING failed to parse beta-factor at line 10111 @> WARNING failed to parse beta-factor at line 10112 @> WARNING failed to parse beta-factor at line 10113 @> WARNING failed to parse beta-factor at line 10114 @> WARNING failed to parse beta-factor at line 10115 @> WARNING failed to parse beta-factor at line 10116 @> WARNING failed to parse beta-factor at line 10117 @> WARNING failed to parse beta-factor at line 10118 @> WARNING failed to parse beta-factor at line 10119 @> WARNING failed to parse beta-factor at line 10120 @> WARNING failed to parse beta-factor at line 10121 @> WARNING failed to parse beta-factor at line 10122 @> WARNING failed to parse beta-factor at line 10123 @> WARNING failed to parse beta-factor at line 10124 @> WARNING failed to parse beta-factor at line 10125 @> WARNING failed to parse beta-factor at line 10126 @> WARNING failed to parse beta-factor at line 10127 @> WARNING failed to parse beta-factor at line 10128 @> WARNING failed to parse beta-factor at line 10129 @> WARNING failed to parse beta-factor at line 10130 @> WARNING failed to parse beta-factor at line 10131 @> WARNING failed to parse beta-factor at line 10132 @> WARNING failed to parse beta-factor at line 10133 @> WARNING failed to parse beta-factor at line 10134 @> WARNING failed to parse beta-factor at line 10135 @> WARNING failed to parse beta-factor at line 10136 @> WARNING failed to parse beta-factor at line 10137 @> WARNING failed to parse beta-factor at line 10138 @> WARNING failed to parse beta-factor at line 10139 @> WARNING failed to parse beta-factor at line 10140 @> WARNING failed to parse beta-factor at line 10141 @> WARNING failed to parse beta-factor at line 10142 @> WARNING failed to parse beta-factor at line 10143 @> WARNING failed to parse beta-factor at line 10144 @> WARNING failed to parse beta-factor at line 10145 @> WARNING failed to parse beta-factor at line 10146 @> WARNING failed to parse beta-factor at line 10147 @> WARNING failed to parse beta-factor at line 10148 @> WARNING failed to parse beta-factor at line 10149 @> WARNING failed to parse beta-factor at line 10150 @> WARNING failed to parse beta-factor at line 10151 @> WARNING failed to parse beta-factor at line 10152 @> WARNING failed to parse beta-factor at line 10153 @> WARNING failed to parse beta-factor at line 10154 @> WARNING failed to parse beta-factor at line 10155 @> WARNING failed to parse beta-factor at line 10156 @> WARNING failed to parse beta-factor at line 10157 @> WARNING failed to parse beta-factor at line 10158 @> WARNING failed to parse beta-factor at line 10159 @> WARNING failed to parse beta-factor at line 10160 @> WARNING failed to parse beta-factor at line 10161 @> WARNING failed to parse beta-factor at line 10162 @> WARNING failed to parse beta-factor at line 10163 @> WARNING failed to parse beta-factor at line 10164 @> WARNING failed to parse beta-factor at line 10165 @> WARNING failed to parse beta-factor at line 10166 @> WARNING failed to parse beta-factor at line 10167 @> WARNING failed to parse beta-factor at line 10168 @> WARNING failed to parse beta-factor at line 10169 @> WARNING failed to parse beta-factor at line 10170 @> WARNING failed to parse beta-factor at line 10171 @> WARNING failed to parse beta-factor at line 10172 @> WARNING failed to parse beta-factor at line 10173 @> WARNING failed to parse beta-factor at line 10174 @> WARNING failed to parse beta-factor at line 10175 @> WARNING failed to parse beta-factor at line 10176 @> WARNING failed to parse beta-factor at line 10177 @> WARNING failed to parse beta-factor at line 10178 @> WARNING failed to parse beta-factor at line 10179 @> WARNING failed to parse beta-factor at line 10180 @> WARNING failed to parse beta-factor at line 10181 @> WARNING failed to parse beta-factor at line 10182 @> WARNING failed to parse beta-factor at line 10183 @> WARNING failed to parse beta-factor at line 10184 @> WARNING failed to parse beta-factor at line 10185 @> WARNING failed to parse beta-factor at line 10186 @> WARNING failed to parse beta-factor at line 10187 @> WARNING failed to parse beta-factor at line 10188 @> WARNING failed to parse beta-factor at line 10189 @> WARNING failed to parse beta-factor at line 10190 @> WARNING failed to parse beta-factor at line 10191 @> WARNING failed to parse beta-factor at line 10192 @> WARNING failed to parse beta-factor at line 10193 @> WARNING failed to parse beta-factor at line 10194 @> WARNING failed to parse beta-factor at line 10195 @> WARNING failed to parse beta-factor at line 10196 @> WARNING failed to parse beta-factor at line 10197 @> WARNING failed to parse beta-factor at line 10198 @> WARNING failed to parse beta-factor at line 10199 @> WARNING failed to parse beta-factor at line 10200 @> WARNING failed to parse beta-factor at line 10201 @> WARNING failed to parse beta-factor at line 10202 @> WARNING failed to parse beta-factor at line 10203 @> WARNING failed to parse beta-factor at line 10204 @> WARNING failed to parse beta-factor at line 10205 @> WARNING failed to parse beta-factor at line 10206 @> WARNING failed to parse beta-factor at line 10207 @> WARNING failed to parse beta-factor at line 10208 @> WARNING failed to parse beta-factor at line 10209 @> WARNING failed to parse beta-factor at line 10210 @> WARNING failed to parse beta-factor at line 10211 @> WARNING failed to parse beta-factor at line 10212 @> WARNING failed to parse beta-factor at line 10213 @> WARNING failed to parse beta-factor at line 10214 @> WARNING failed to parse beta-factor at line 10215 @> WARNING failed to parse beta-factor at line 10216 @> WARNING failed to parse beta-factor at line 10217 @> WARNING failed to parse beta-factor at line 10218 @> WARNING failed to parse beta-factor at line 10219 @> WARNING failed to parse beta-factor at line 10220 @> WARNING failed to parse beta-factor at line 10221 @> WARNING failed to parse beta-factor at line 10222 @> WARNING failed to parse beta-factor at line 10223 @> WARNING failed to parse beta-factor at line 10224 @> WARNING failed to parse beta-factor at line 10225 @> WARNING failed to parse beta-factor at line 10226 @> WARNING failed to parse beta-factor at line 10227 @> WARNING failed to parse beta-factor at line 10228 @> WARNING failed to parse beta-factor at line 10229 @> WARNING failed to parse beta-factor at line 10230 @> WARNING failed to parse beta-factor at line 10231 @> WARNING failed to parse beta-factor at line 10232 @> WARNING failed to parse beta-factor at line 10233 @> WARNING failed to parse beta-factor at line 10234 @> WARNING failed to parse beta-factor at line 10235 @> WARNING failed to parse beta-factor at line 10236 @> WARNING failed to parse beta-factor at line 10237 @> WARNING failed to parse beta-factor at line 10238 @> WARNING failed to parse beta-factor at line 10239 @> WARNING failed to parse beta-factor at line 10240 @> WARNING failed to parse beta-factor at line 10241 @> WARNING failed to parse beta-factor at line 10242 @> WARNING failed to parse beta-factor at line 10243 @> WARNING failed to parse beta-factor at line 10244 @> WARNING failed to parse beta-factor at line 10245 @> WARNING failed to parse beta-factor at line 10246 @> WARNING failed to parse beta-factor at line 10247 @> WARNING failed to parse beta-factor at line 10248 @> WARNING failed to parse beta-factor at line 10249 @> WARNING failed to parse beta-factor at line 10250 @> WARNING failed to parse beta-factor at line 10251 @> WARNING failed to parse beta-factor at line 10252 @> WARNING failed to parse beta-factor at line 10253 @> WARNING failed to parse beta-factor at line 10254 @> WARNING failed to parse beta-factor at line 10255 @> WARNING failed to parse beta-factor at line 10256 @> WARNING failed to parse beta-factor at line 10257 @> WARNING failed to parse beta-factor at line 10258 @> WARNING failed to parse beta-factor at line 10259 @> WARNING failed to parse beta-factor at line 10260 @> WARNING failed to parse beta-factor at line 10261 @> WARNING failed to parse beta-factor at line 10262 @> WARNING failed to parse beta-factor at line 10263 @> WARNING failed to parse beta-factor at line 10264 @> WARNING failed to parse beta-factor at line 10265 @> WARNING failed to parse beta-factor at line 10266 @> WARNING failed to parse beta-factor at line 10267 @> WARNING failed to parse beta-factor at line 10268 @> WARNING failed to parse beta-factor at line 10269 @> WARNING failed to parse beta-factor at line 10270 @> WARNING failed to parse beta-factor at line 10271 @> WARNING failed to parse beta-factor at line 10272 @> WARNING failed to parse beta-factor at line 10273 @> WARNING failed to parse beta-factor at line 10274 @> WARNING failed to parse beta-factor at line 10275 @> WARNING failed to parse beta-factor at line 10276 @> WARNING failed to parse beta-factor at line 10277 @> WARNING failed to parse beta-factor at line 10278 @> WARNING failed to parse beta-factor at line 10279 @> WARNING failed to parse beta-factor at line 10280 @> WARNING failed to parse beta-factor at line 10281 @> WARNING failed to parse beta-factor at line 10282 @> WARNING failed to parse beta-factor at line 10283 @> WARNING failed to parse beta-factor at line 10284 @> WARNING failed to parse beta-factor at line 10285 @> WARNING failed to parse beta-factor at line 10286 @> WARNING failed to parse beta-factor at line 10287 @> WARNING failed to parse beta-factor at line 10288 @> WARNING failed to parse beta-factor at line 10289 @> WARNING failed to parse beta-factor at line 10290 @> WARNING failed to parse beta-factor at line 10291 @> WARNING failed to parse beta-factor at line 10292 @> WARNING failed to parse beta-factor at line 10293 @> WARNING failed to parse beta-factor at line 10294 @> WARNING failed to parse beta-factor at line 10295 @> WARNING failed to parse beta-factor at line 10296 @> WARNING failed to parse beta-factor at line 10297 @> WARNING failed to parse beta-factor at line 10298 @> WARNING failed to parse beta-factor at line 10299 @> WARNING failed to parse beta-factor at line 10300 @> WARNING failed to parse beta-factor at line 10301 @> WARNING failed to parse beta-factor at line 10302 @> WARNING failed to parse beta-factor at line 10303 @> WARNING failed to parse beta-factor at line 10304 @> WARNING failed to parse beta-factor at line 10305 @> WARNING failed to parse beta-factor at line 10306 @> WARNING failed to parse beta-factor at line 10307 @> WARNING failed to parse beta-factor at line 10308 @> WARNING failed to parse beta-factor at line 10309 @> WARNING failed to parse beta-factor at line 10310 @> WARNING failed to parse beta-factor at line 10311 @> WARNING failed to parse beta-factor at line 10312 @> WARNING failed to parse beta-factor at line 10313 @> WARNING failed to parse beta-factor at line 10314 @> WARNING failed to parse beta-factor at line 10315 @> WARNING failed to parse beta-factor at line 10316 @> WARNING failed to parse beta-factor at line 10317 @> WARNING failed to parse beta-factor at line 10318 @> WARNING failed to parse beta-factor at line 10319 @> WARNING failed to parse beta-factor at line 10320 @> WARNING failed to parse beta-factor at line 10321 @> WARNING failed to parse beta-factor at line 10322 @> WARNING failed to parse beta-factor at line 10323 @> WARNING failed to parse beta-factor at line 10324 @> WARNING failed to parse beta-factor at line 10325 @> WARNING failed to parse beta-factor at line 10326 @> WARNING failed to parse beta-factor at line 10327 @> WARNING failed to parse beta-factor at line 10328 @> WARNING failed to parse beta-factor at line 10329 @> WARNING failed to parse beta-factor at line 10330 @> WARNING failed to parse beta-factor at line 10331 @> WARNING failed to parse beta-factor at line 10332 @> WARNING failed to parse beta-factor at line 10333 @> WARNING failed to parse beta-factor at line 10334 @> WARNING failed to parse beta-factor at line 10335 @> WARNING failed to parse beta-factor at line 10336 @> WARNING failed to parse beta-factor at line 10337 @> WARNING failed to parse beta-factor at line 10338 @> WARNING failed to parse beta-factor at line 10339 @> WARNING failed to parse beta-factor at line 10340 @> WARNING failed to parse beta-factor at line 10341 @> WARNING failed to parse beta-factor at line 10342 @> WARNING failed to parse beta-factor at line 10343 @> WARNING failed to parse beta-factor at line 10344 @> WARNING failed to parse beta-factor at line 10345 @> WARNING failed to parse beta-factor at line 10346 @> WARNING failed to parse beta-factor at line 10347 @> WARNING failed to parse beta-factor at line 10348 @> WARNING failed to parse beta-factor at line 10349 @> WARNING failed to parse beta-factor at line 10350 @> WARNING failed to parse beta-factor at line 10351 @> WARNING failed to parse beta-factor at line 10352 @> WARNING failed to parse beta-factor at line 10353 @> WARNING failed to parse beta-factor at line 10354 @> WARNING failed to parse beta-factor at line 10355 @> WARNING failed to parse beta-factor at line 10356 @> WARNING failed to parse beta-factor at line 10357 @> WARNING failed to parse beta-factor at line 10358 @> WARNING failed to parse beta-factor at line 10359 @> WARNING failed to parse beta-factor at line 10360 @> WARNING failed to parse beta-factor at line 10361 @> WARNING failed to parse beta-factor at line 10362 @> WARNING failed to parse beta-factor at line 10363 @> WARNING failed to parse beta-factor at line 10364 @> WARNING failed to parse beta-factor at line 10365 @> WARNING failed to parse beta-factor at line 10366 @> WARNING failed to parse beta-factor at line 10367 @> WARNING failed to parse beta-factor at line 10368 @> WARNING failed to parse beta-factor at line 10369 @> WARNING failed to parse beta-factor at line 10370 @> WARNING failed to parse beta-factor at line 10371 @> WARNING failed to parse beta-factor at line 10372 @> WARNING failed to parse beta-factor at line 10373 @> WARNING failed to parse beta-factor at line 10374 @> WARNING failed to parse beta-factor at line 10375 @> WARNING failed to parse beta-factor at line 10376 @> WARNING failed to parse beta-factor at line 10377 @> WARNING failed to parse beta-factor at line 10378 @> WARNING failed to parse beta-factor at line 10379 @> WARNING failed to parse beta-factor at line 10380 @> WARNING failed to parse beta-factor at line 10381 @> WARNING failed to parse beta-factor at line 10382 @> WARNING failed to parse beta-factor at line 10383 @> WARNING failed to parse beta-factor at line 10384 @> WARNING failed to parse beta-factor at line 10385 @> WARNING failed to parse beta-factor at line 10386 @> WARNING failed to parse beta-factor at line 10387 @> WARNING failed to parse beta-factor at line 10388 @> WARNING failed to parse beta-factor at line 10389 @> WARNING failed to parse beta-factor at line 10390 @> WARNING failed to parse beta-factor at line 10391 @> WARNING failed to parse beta-factor at line 10392 @> WARNING failed to parse beta-factor at line 10393 @> WARNING failed to parse beta-factor at line 10394 @> WARNING failed to parse beta-factor at line 10395 @> WARNING failed to parse beta-factor at line 10396 @> WARNING failed to parse beta-factor at line 10397 @> WARNING failed to parse beta-factor at line 10398 @> WARNING failed to parse beta-factor at line 10399 @> WARNING failed to parse beta-factor at line 10400 @> WARNING failed to parse beta-factor at line 10401 @> WARNING failed to parse beta-factor at line 10402 @> WARNING failed to parse beta-factor at line 10403 @> WARNING failed to parse beta-factor at line 10404 @> WARNING failed to parse beta-factor at line 10405 @> WARNING failed to parse beta-factor at line 10406 @> WARNING failed to parse beta-factor at line 10407 @> WARNING failed to parse beta-factor at line 10408 @> WARNING failed to parse beta-factor at line 10409 @> WARNING failed to parse beta-factor at line 10410 @> WARNING failed to parse beta-factor at line 10411 @> WARNING failed to parse beta-factor at line 10412 @> WARNING failed to parse beta-factor at line 10413 @> WARNING failed to parse beta-factor at line 10414 @> WARNING failed to parse beta-factor at line 10415 @> WARNING failed to parse beta-factor at line 10416 @> WARNING failed to parse beta-factor at line 10417 @> WARNING failed to parse beta-factor at line 10418 @> WARNING failed to parse beta-factor at line 10419 @> WARNING failed to parse beta-factor at line 10420 @> WARNING failed to parse beta-factor at line 10421 @> WARNING failed to parse beta-factor at line 10422 @> WARNING failed to parse beta-factor at line 10423 @> WARNING failed to parse beta-factor at line 10424 @> WARNING failed to parse beta-factor at line 10425 @> WARNING failed to parse beta-factor at line 10426 @> WARNING failed to parse beta-factor at line 10427 @> WARNING failed to parse beta-factor at line 10428 @> WARNING failed to parse beta-factor at line 10429 @> WARNING failed to parse beta-factor at line 10430 @> WARNING failed to parse beta-factor at line 10431 @> WARNING failed to parse beta-factor at line 10432 @> WARNING failed to parse beta-factor at line 10433 @> WARNING failed to parse beta-factor at line 10434 @> WARNING failed to parse beta-factor at line 10435 @> WARNING failed to parse beta-factor at line 10436 @> WARNING failed to parse beta-factor at line 10437 @> WARNING failed to parse beta-factor at line 10438 @> WARNING failed to parse beta-factor at line 10439 @> WARNING failed to parse beta-factor at line 10440 @> WARNING failed to parse beta-factor at line 10441 @> WARNING failed to parse beta-factor at line 10442 @> WARNING failed to parse beta-factor at line 10443 @> WARNING failed to parse beta-factor at line 10444 @> WARNING failed to parse beta-factor at line 10445 @> WARNING failed to parse beta-factor at line 10446 @> WARNING failed to parse beta-factor at line 10447 @> WARNING failed to parse beta-factor at line 10448 @> WARNING failed to parse beta-factor at line 10449 @> WARNING failed to parse beta-factor at line 10450 @> WARNING failed to parse beta-factor at line 10451 @> WARNING failed to parse beta-factor at line 10452 @> WARNING failed to parse beta-factor at line 10453 @> WARNING failed to parse beta-factor at line 10454 @> WARNING failed to parse beta-factor at line 10455 @> WARNING failed to parse beta-factor at line 10456 @> WARNING failed to parse beta-factor at line 10457 @> WARNING failed to parse beta-factor at line 10458 @> WARNING failed to parse beta-factor at line 10459 @> WARNING failed to parse beta-factor at line 10460 @> WARNING failed to parse beta-factor at line 10461 @> WARNING failed to parse beta-factor at line 10462 @> WARNING failed to parse beta-factor at line 10463 @> WARNING failed to parse beta-factor at line 10464 @> WARNING failed to parse beta-factor at line 10465 @> WARNING failed to parse beta-factor at line 10466 @> WARNING failed to parse beta-factor at line 10467 @> WARNING failed to parse beta-factor at line 10468 @> WARNING failed to parse beta-factor at line 10469 @> WARNING failed to parse beta-factor at line 10470 @> WARNING failed to parse beta-factor at line 10471 @> WARNING failed to parse beta-factor at line 10472 @> WARNING failed to parse beta-factor at line 10473 @> WARNING failed to parse beta-factor at line 10474 @> WARNING failed to parse beta-factor at line 10475 @> WARNING failed to parse beta-factor at line 10476 @> WARNING failed to parse beta-factor at line 10477 @> WARNING failed to parse beta-factor at line 10478 @> WARNING failed to parse beta-factor at line 10479 @> WARNING failed to parse beta-factor at line 10480 @> WARNING failed to parse beta-factor at line 10481 @> WARNING failed to parse beta-factor at line 10482 @> WARNING failed to parse beta-factor at line 10483 @> WARNING failed to parse beta-factor at line 10484 @> WARNING failed to parse beta-factor at line 10485 @> WARNING failed to parse beta-factor at line 10486 @> WARNING failed to parse beta-factor at line 10487 @> WARNING failed to parse beta-factor at line 10488 @> WARNING failed to parse beta-factor at line 10489 @> WARNING failed to parse beta-factor at line 10490 @> WARNING failed to parse beta-factor at line 10491 @> WARNING failed to parse beta-factor at line 10492 @> WARNING failed to parse beta-factor at line 10493 @> WARNING failed to parse beta-factor at line 10494 @> WARNING failed to parse beta-factor at line 10495 @> WARNING failed to parse beta-factor at line 10496 @> WARNING failed to parse beta-factor at line 10497 @> WARNING failed to parse beta-factor at line 10498 @> WARNING failed to parse beta-factor at line 10499 @> WARNING failed to parse beta-factor at line 10500 @> WARNING failed to parse beta-factor at line 10501 @> WARNING failed to parse beta-factor at line 10502 @> WARNING failed to parse beta-factor at line 10503 @> WARNING failed to parse beta-factor at line 10504 @> WARNING failed to parse beta-factor at line 10505 @> WARNING failed to parse beta-factor at line 10506 @> WARNING failed to parse beta-factor at line 10507 @> WARNING failed to parse beta-factor at line 10508 @> WARNING failed to parse beta-factor at line 10509 @> WARNING failed to parse beta-factor at line 10510 @> WARNING failed to parse beta-factor at line 10511 @> WARNING failed to parse beta-factor at line 10512 @> WARNING failed to parse beta-factor at line 10513 @> WARNING failed to parse beta-factor at line 10514 @> WARNING failed to parse beta-factor at line 10515 @> WARNING failed to parse beta-factor at line 10516 @> WARNING failed to parse beta-factor at line 10517 @> WARNING failed to parse beta-factor at line 10518 @> WARNING failed to parse beta-factor at line 10519 @> WARNING failed to parse beta-factor at line 10520 @> WARNING failed to parse beta-factor at line 10521 @> WARNING failed to parse beta-factor at line 10522 @> WARNING failed to parse beta-factor at line 10523 @> WARNING failed to parse beta-factor at line 10524 @> WARNING failed to parse beta-factor at line 10525 @> WARNING failed to parse beta-factor at line 10526 @> WARNING failed to parse beta-factor at line 10527 @> WARNING failed to parse beta-factor at line 10528 @> WARNING failed to parse beta-factor at line 10529 @> WARNING failed to parse beta-factor at line 10530 @> WARNING failed to parse beta-factor at line 10531 @> WARNING failed to parse beta-factor at line 10532 @> WARNING failed to parse beta-factor at line 10533 @> WARNING failed to parse beta-factor at line 10534 @> WARNING failed to parse beta-factor at line 10535 @> WARNING failed to parse beta-factor at line 10536 @> WARNING failed to parse beta-factor at line 10537 @> WARNING failed to parse beta-factor at line 10538 @> WARNING failed to parse beta-factor at line 10539 @> WARNING failed to parse beta-factor at line 10540 @> WARNING failed to parse beta-factor at line 10541 @> WARNING failed to parse beta-factor at line 10542 @> WARNING failed to parse beta-factor at line 10543 @> WARNING failed to parse beta-factor at line 10544 @> WARNING failed to parse beta-factor at line 10545 @> WARNING failed to parse beta-factor at line 10546 @> WARNING failed to parse beta-factor at line 10547 @> WARNING failed to parse beta-factor at line 10548 @> WARNING failed to parse beta-factor at line 10549 @> WARNING failed to parse beta-factor at line 10550 @> WARNING failed to parse beta-factor at line 10551 @> WARNING failed to parse beta-factor at line 10552 @> WARNING failed to parse beta-factor at line 10553 @> WARNING failed to parse beta-factor at line 10554 @> WARNING failed to parse beta-factor at line 10555 @> WARNING failed to parse beta-factor at line 10556 @> WARNING failed to parse beta-factor at line 10557 @> WARNING failed to parse beta-factor at line 10558 @> WARNING failed to parse beta-factor at line 10559 @> WARNING failed to parse beta-factor at line 10560 @> WARNING failed to parse beta-factor at line 10561 @> WARNING failed to parse beta-factor at line 10562 @> WARNING failed to parse beta-factor at line 10563 @> WARNING failed to parse beta-factor at line 10564 @> WARNING failed to parse beta-factor at line 10565 @> WARNING failed to parse beta-factor at line 10566 @> WARNING failed to parse beta-factor at line 10567 @> WARNING failed to parse beta-factor at line 10568 @> WARNING failed to parse beta-factor at line 10569 @> WARNING failed to parse beta-factor at line 10570 @> WARNING failed to parse beta-factor at line 10571 @> WARNING failed to parse beta-factor at line 10572 @> WARNING failed to parse beta-factor at line 10573 @> WARNING failed to parse beta-factor at line 10574 @> WARNING failed to parse beta-factor at line 10575 @> WARNING failed to parse beta-factor at line 10576 @> WARNING failed to parse beta-factor at line 10577 @> WARNING failed to parse beta-factor at line 10578 @> WARNING failed to parse beta-factor at line 10579 @> WARNING failed to parse beta-factor at line 10580 @> WARNING failed to parse beta-factor at line 10581 @> WARNING failed to parse beta-factor at line 10582 @> WARNING failed to parse beta-factor at line 10583 @> WARNING failed to parse beta-factor at line 10584 @> WARNING failed to parse beta-factor at line 10585 @> WARNING failed to parse beta-factor at line 10586 @> WARNING failed to parse beta-factor at line 10587 @> WARNING failed to parse beta-factor at line 10588 @> WARNING failed to parse beta-factor at line 10589 @> WARNING failed to parse beta-factor at line 10590 @> WARNING failed to parse beta-factor at line 10591 @> WARNING failed to parse beta-factor at line 10592 @> WARNING failed to parse beta-factor at line 10593 @> WARNING failed to parse beta-factor at line 10594 @> WARNING failed to parse beta-factor at line 10595 @> WARNING failed to parse beta-factor at line 10596 @> WARNING failed to parse beta-factor at line 10597 @> WARNING failed to parse beta-factor at line 10598 @> WARNING failed to parse beta-factor at line 10599 @> WARNING failed to parse beta-factor at line 10600 @> WARNING failed to parse beta-factor at line 10601 @> WARNING failed to parse beta-factor at line 10602 @> WARNING failed to parse beta-factor at line 10603 @> WARNING failed to parse beta-factor at line 10604 @> WARNING failed to parse beta-factor at line 10605 @> WARNING failed to parse beta-factor at line 10606 @> WARNING failed to parse beta-factor at line 10607 @> WARNING failed to parse beta-factor at line 10608 @> WARNING failed to parse beta-factor at line 10609 @> WARNING failed to parse beta-factor at line 10610 @> WARNING failed to parse beta-factor at line 10611 @> WARNING failed to parse beta-factor at line 10612 @> WARNING failed to parse beta-factor at line 10613 @> WARNING failed to parse beta-factor at line 10614 @> WARNING failed to parse beta-factor at line 10615 @> WARNING failed to parse beta-factor at line 10616 @> WARNING failed to parse beta-factor at line 10617 @> WARNING failed to parse beta-factor at line 10618 @> WARNING failed to parse beta-factor at line 10619 @> WARNING failed to parse beta-factor at line 10620 @> WARNING failed to parse beta-factor at line 10621 @> WARNING failed to parse beta-factor at line 10622 @> WARNING failed to parse beta-factor at line 10623 @> WARNING failed to parse beta-factor at line 10624 @> WARNING failed to parse beta-factor at line 10625 @> WARNING failed to parse beta-factor at line 10626 @> WARNING failed to parse beta-factor at line 10627 @> WARNING failed to parse beta-factor at line 10628 @> WARNING failed to parse beta-factor at line 10629 @> WARNING failed to parse beta-factor at line 10630 @> WARNING failed to parse beta-factor at line 10631 @> WARNING failed to parse beta-factor at line 10632 @> WARNING failed to parse beta-factor at line 10633 @> WARNING failed to parse beta-factor at line 10634 @> WARNING failed to parse beta-factor at line 10635 @> WARNING failed to parse beta-factor at line 10636 @> WARNING failed to parse beta-factor at line 10637 @> WARNING failed to parse beta-factor at line 10638 @> WARNING failed to parse beta-factor at line 10639 @> WARNING failed to parse beta-factor at line 10640 @> WARNING failed to parse beta-factor at line 10641 @> WARNING failed to parse beta-factor at line 10642 @> WARNING failed to parse beta-factor at line 10643 @> WARNING failed to parse beta-factor at line 10644 @> WARNING failed to parse beta-factor at line 10645 @> WARNING failed to parse beta-factor at line 10646 @> WARNING failed to parse beta-factor at line 10647 @> WARNING failed to parse beta-factor at line 10648 @> WARNING failed to parse beta-factor at line 10649 @> WARNING failed to parse beta-factor at line 10650 @> WARNING failed to parse beta-factor at line 10651 @> WARNING failed to parse beta-factor at line 10652 @> WARNING failed to parse beta-factor at line 10653 @> WARNING failed to parse beta-factor at line 10654 @> WARNING failed to parse beta-factor at line 10655 @> WARNING failed to parse beta-factor at line 10656 @> WARNING failed to parse beta-factor at line 10657 @> WARNING failed to parse beta-factor at line 10658 @> WARNING failed to parse beta-factor at line 10659 @> WARNING failed to parse beta-factor at line 10660 @> WARNING failed to parse beta-factor at line 10661 @> WARNING failed to parse beta-factor at line 10662 @> WARNING failed to parse beta-factor at line 10663 @> WARNING failed to parse beta-factor at line 10664 @> WARNING failed to parse beta-factor at line 10665 @> WARNING failed to parse beta-factor at line 10666 @> WARNING failed to parse beta-factor at line 10667 @> WARNING failed to parse beta-factor at line 10668 @> WARNING failed to parse beta-factor at line 10669 @> WARNING failed to parse beta-factor at line 10670 @> WARNING failed to parse beta-factor at line 10671 @> WARNING failed to parse beta-factor at line 10672 @> WARNING failed to parse beta-factor at line 10673 @> WARNING failed to parse beta-factor at line 10674 @> WARNING failed to parse beta-factor at line 10675 @> WARNING failed to parse beta-factor at line 10676 @> WARNING failed to parse beta-factor at line 10677 @> WARNING failed to parse beta-factor at line 10678 @> WARNING failed to parse beta-factor at line 10679 @> WARNING failed to parse beta-factor at line 10680 @> WARNING failed to parse beta-factor at line 10681 @> WARNING failed to parse beta-factor at line 10682 @> WARNING failed to parse beta-factor at line 10683 @> WARNING failed to parse beta-factor at line 10684 @> WARNING failed to parse beta-factor at line 10685 @> WARNING failed to parse beta-factor at line 10686 @> WARNING failed to parse beta-factor at line 10687 @> WARNING failed to parse beta-factor at line 10688 @> WARNING failed to parse beta-factor at line 10689 @> WARNING failed to parse beta-factor at line 10690 @> WARNING failed to parse beta-factor at line 10691 @> WARNING failed to parse beta-factor at line 10692 @> WARNING failed to parse beta-factor at line 10693 @> WARNING failed to parse beta-factor at line 10694 @> WARNING failed to parse beta-factor at line 10695 @> WARNING failed to parse beta-factor at line 10696 @> WARNING failed to parse beta-factor at line 10697 @> WARNING failed to parse beta-factor at line 10698 @> WARNING failed to parse beta-factor at line 10699 @> WARNING failed to parse beta-factor at line 10700 @> WARNING failed to parse beta-factor at line 10701 @> WARNING failed to parse beta-factor at line 10702 @> WARNING failed to parse beta-factor at line 10703 @> WARNING failed to parse beta-factor at line 10704 @> WARNING failed to parse beta-factor at line 10705 @> WARNING failed to parse beta-factor at line 10706 @> WARNING failed to parse beta-factor at line 10707 @> WARNING failed to parse beta-factor at line 10708 @> WARNING failed to parse beta-factor at line 10709 @> WARNING failed to parse beta-factor at line 10710 @> WARNING failed to parse beta-factor at line 10711 @> WARNING failed to parse beta-factor at line 10712 @> WARNING failed to parse beta-factor at line 10713 @> WARNING failed to parse beta-factor at line 10714 @> WARNING failed to parse beta-factor at line 10715 @> WARNING failed to parse beta-factor at line 10716 @> WARNING failed to parse beta-factor at line 10717 @> WARNING failed to parse beta-factor at line 10718 @> WARNING failed to parse beta-factor at line 10719 @> WARNING failed to parse beta-factor at line 10720 @> WARNING failed to parse beta-factor at line 10721 @> WARNING failed to parse beta-factor at line 10722 @> WARNING failed to parse beta-factor at line 10723 @> WARNING failed to parse beta-factor at line 10724 @> WARNING failed to parse beta-factor at line 10725 @> WARNING failed to parse beta-factor at line 10726 @> WARNING failed to parse beta-factor at line 10727 @> WARNING failed to parse beta-factor at line 10728 @> WARNING failed to parse beta-factor at line 10729 @> WARNING failed to parse beta-factor at line 10730 @> WARNING failed to parse beta-factor at line 10731 @> WARNING failed to parse beta-factor at line 10732 @> WARNING failed to parse beta-factor at line 10733 @> WARNING failed to parse beta-factor at line 10734 @> WARNING failed to parse beta-factor at line 10735 @> WARNING failed to parse beta-factor at line 10736 @> WARNING failed to parse beta-factor at line 10737 @> WARNING failed to parse beta-factor at line 10738 @> WARNING failed to parse beta-factor at line 10739 @> WARNING failed to parse beta-factor at line 10740 @> WARNING failed to parse beta-factor at line 10741 @> WARNING failed to parse beta-factor at line 10742 @> WARNING failed to parse beta-factor at line 10743 @> WARNING failed to parse beta-factor at line 10744 @> WARNING failed to parse beta-factor at line 10745 @> WARNING failed to parse beta-factor at line 10746 @> WARNING failed to parse beta-factor at line 10747 @> WARNING failed to parse beta-factor at line 10748 @> WARNING failed to parse beta-factor at line 10749 @> WARNING failed to parse beta-factor at line 10750 @> WARNING failed to parse beta-factor at line 10751 @> WARNING failed to parse beta-factor at line 10752 @> WARNING failed to parse beta-factor at line 10753 @> WARNING failed to parse beta-factor at line 10754 @> WARNING failed to parse beta-factor at line 10755 @> WARNING failed to parse beta-factor at line 10756 @> WARNING failed to parse beta-factor at line 10757 @> WARNING failed to parse beta-factor at line 10758 @> WARNING failed to parse beta-factor at line 10759 @> WARNING failed to parse beta-factor at line 10760 @> WARNING failed to parse beta-factor at line 10761 @> WARNING failed to parse beta-factor at line 10762 @> WARNING failed to parse beta-factor at line 10763 @> WARNING failed to parse beta-factor at line 10764 @> WARNING failed to parse beta-factor at line 10765 @> WARNING failed to parse beta-factor at line 10766 @> WARNING failed to parse beta-factor at line 10767 @> WARNING failed to parse beta-factor at line 10768 @> WARNING failed to parse beta-factor at line 10769 @> WARNING failed to parse beta-factor at line 10770 @> WARNING failed to parse beta-factor at line 10771 @> WARNING failed to parse beta-factor at line 10772 @> WARNING failed to parse beta-factor at line 10773 @> WARNING failed to parse beta-factor at line 10774 @> WARNING failed to parse beta-factor at line 10775 @> WARNING failed to parse beta-factor at line 10776 @> WARNING failed to parse beta-factor at line 10777 @> WARNING failed to parse beta-factor at line 10778 @> WARNING failed to parse beta-factor at line 10779 @> WARNING failed to parse beta-factor at line 10780 @> WARNING failed to parse beta-factor at line 10781 @> WARNING failed to parse beta-factor at line 10782 @> WARNING failed to parse beta-factor at line 10783 @> WARNING failed to parse beta-factor at line 10784 @> WARNING failed to parse beta-factor at line 10785 @> WARNING failed to parse beta-factor at line 10786 @> WARNING failed to parse beta-factor at line 10787 @> WARNING failed to parse beta-factor at line 10788 @> WARNING failed to parse beta-factor at line 10789 @> WARNING failed to parse beta-factor at line 10790 @> WARNING failed to parse beta-factor at line 10791 @> WARNING failed to parse beta-factor at line 10792 @> WARNING failed to parse beta-factor at line 10793 @> WARNING failed to parse beta-factor at line 10794 @> WARNING failed to parse beta-factor at line 10795 @> WARNING failed to parse beta-factor at line 10796 @> WARNING failed to parse beta-factor at line 10797 @> WARNING failed to parse beta-factor at line 10798 @> WARNING failed to parse beta-factor at line 10799 @> WARNING failed to parse beta-factor at line 10800 @> WARNING failed to parse beta-factor at line 10801 @> WARNING failed to parse beta-factor at line 10802 @> WARNING failed to parse beta-factor at line 10803 @> WARNING failed to parse beta-factor at line 10804 @> WARNING failed to parse beta-factor at line 10805 @> WARNING failed to parse beta-factor at line 10806 @> WARNING failed to parse beta-factor at line 10807 @> WARNING failed to parse beta-factor at line 10808 @> WARNING failed to parse beta-factor at line 10809 @> WARNING failed to parse beta-factor at line 10810 @> WARNING failed to parse beta-factor at line 10811 @> WARNING failed to parse beta-factor at line 10812 @> WARNING failed to parse beta-factor at line 10813 @> WARNING failed to parse beta-factor at line 10814 @> WARNING failed to parse beta-factor at line 10815 @> WARNING failed to parse beta-factor at line 10816 @> WARNING failed to parse beta-factor at line 10817 @> WARNING failed to parse beta-factor at line 10818 @> WARNING failed to parse beta-factor at line 10819 @> WARNING failed to parse beta-factor at line 10820 @> WARNING failed to parse beta-factor at line 10821 @> WARNING failed to parse beta-factor at line 10822 @> WARNING failed to parse beta-factor at line 10823 @> WARNING failed to parse beta-factor at line 10824 @> WARNING failed to parse beta-factor at line 10825 @> WARNING failed to parse beta-factor at line 10826 @> WARNING failed to parse beta-factor at line 10827 @> WARNING failed to parse beta-factor at line 10828 @> WARNING failed to parse beta-factor at line 10829 @> WARNING failed to parse beta-factor at line 10830 @> WARNING failed to parse beta-factor at line 10831 @> WARNING failed to parse beta-factor at line 10832 @> WARNING failed to parse beta-factor at line 10833 @> WARNING failed to parse beta-factor at line 10834 @> WARNING failed to parse beta-factor at line 10835 @> WARNING failed to parse beta-factor at line 10836 @> WARNING failed to parse beta-factor at line 10837 @> WARNING failed to parse beta-factor at line 10838 @> WARNING failed to parse beta-factor at line 10839 @> WARNING failed to parse beta-factor at line 10840 @> WARNING failed to parse beta-factor at line 10841 @> WARNING failed to parse beta-factor at line 10842 @> WARNING failed to parse beta-factor at line 10843 @> WARNING failed to parse beta-factor at line 10844 @> WARNING failed to parse beta-factor at line 10845 @> WARNING failed to parse beta-factor at line 10846 @> WARNING failed to parse beta-factor at line 10847 @> WARNING failed to parse beta-factor at line 10848 @> WARNING failed to parse beta-factor at line 10849 @> WARNING failed to parse beta-factor at line 10850 @> WARNING failed to parse beta-factor at line 10851 @> WARNING failed to parse beta-factor at line 10852 @> WARNING failed to parse beta-factor at line 10853 @> WARNING failed to parse beta-factor at line 10854 @> WARNING failed to parse beta-factor at line 10855 @> WARNING failed to parse beta-factor at line 10856 @> WARNING failed to parse beta-factor at line 10857 @> WARNING failed to parse beta-factor at line 10858 @> WARNING failed to parse beta-factor at line 10859 @> WARNING failed to parse beta-factor at line 10860 @> WARNING failed to parse beta-factor at line 10861 @> WARNING failed to parse beta-factor at line 10862 @> WARNING failed to parse beta-factor at line 10863 @> WARNING failed to parse beta-factor at line 10864 @> WARNING failed to parse beta-factor at line 10865 @> WARNING failed to parse beta-factor at line 10866 @> WARNING failed to parse beta-factor at line 10867 @> WARNING failed to parse beta-factor at line 10868 @> WARNING failed to parse beta-factor at line 10869 @> WARNING failed to parse beta-factor at line 10870 @> WARNING failed to parse beta-factor at line 10871 @> WARNING failed to parse beta-factor at line 10872 @> WARNING failed to parse beta-factor at line 10873 @> WARNING failed to parse beta-factor at line 10874 @> WARNING failed to parse beta-factor at line 10875 @> WARNING failed to parse beta-factor at line 10876 @> WARNING failed to parse beta-factor at line 10877 @> WARNING failed to parse beta-factor at line 10878 @> WARNING failed to parse beta-factor at line 10879 @> WARNING failed to parse beta-factor at line 10880 @> WARNING failed to parse beta-factor at line 10881 @> WARNING failed to parse beta-factor at line 10882 @> WARNING failed to parse beta-factor at line 10883 @> WARNING failed to parse beta-factor at line 10884 @> WARNING failed to parse beta-factor at line 10885 @> WARNING failed to parse beta-factor at line 10886 @> WARNING failed to parse beta-factor at line 10887 @> WARNING failed to parse beta-factor at line 10888 @> WARNING failed to parse beta-factor at line 10889 @> WARNING failed to parse beta-factor at line 10890 @> WARNING failed to parse beta-factor at line 10891 @> WARNING failed to parse beta-factor at line 10892 @> WARNING failed to parse beta-factor at line 10893 @> WARNING failed to parse beta-factor at line 10894 @> WARNING failed to parse beta-factor at line 10895 @> WARNING failed to parse beta-factor at line 10896 @> WARNING failed to parse beta-factor at line 10897 @> WARNING failed to parse beta-factor at line 10898 @> WARNING failed to parse beta-factor at line 10899 @> WARNING failed to parse beta-factor at line 10900 @> WARNING failed to parse beta-factor at line 10901 @> WARNING failed to parse beta-factor at line 10902 @> WARNING failed to parse beta-factor at line 10903 @> WARNING failed to parse beta-factor at line 10904 @> WARNING failed to parse beta-factor at line 10905 @> WARNING failed to parse beta-factor at line 10906 @> WARNING failed to parse beta-factor at line 10907 @> WARNING failed to parse beta-factor at line 10908 @> WARNING failed to parse beta-factor at line 10909 @> WARNING failed to parse beta-factor at line 10910 @> WARNING failed to parse beta-factor at line 10911 @> WARNING failed to parse beta-factor at line 10912 @> WARNING failed to parse beta-factor at line 10913 @> WARNING failed to parse beta-factor at line 10914 @> WARNING failed to parse beta-factor at line 10915 @> WARNING failed to parse beta-factor at line 10916 @> WARNING failed to parse beta-factor at line 10917 @> WARNING failed to parse beta-factor at line 10918 @> WARNING failed to parse beta-factor at line 10919 @> WARNING failed to parse beta-factor at line 10920 @> WARNING failed to parse beta-factor at line 10921 @> WARNING failed to parse beta-factor at line 10922 @> WARNING failed to parse beta-factor at line 10923 @> WARNING failed to parse beta-factor at line 10924 @> WARNING failed to parse beta-factor at line 10925 @> WARNING failed to parse beta-factor at line 10926 @> WARNING failed to parse beta-factor at line 10927 @> WARNING failed to parse beta-factor at line 10928 @> WARNING failed to parse beta-factor at line 10929 @> WARNING failed to parse beta-factor at line 10930 @> WARNING failed to parse beta-factor at line 10931 @> WARNING failed to parse beta-factor at line 10932 @> WARNING failed to parse beta-factor at line 10933 @> WARNING failed to parse beta-factor at line 10934 @> WARNING failed to parse beta-factor at line 10935 @> WARNING failed to parse beta-factor at line 10936 @> WARNING failed to parse beta-factor at line 10937 @> WARNING failed to parse beta-factor at line 10938 @> WARNING failed to parse beta-factor at line 10939 @> WARNING failed to parse beta-factor at line 10940 @> WARNING failed to parse beta-factor at line 10941 @> WARNING failed to parse beta-factor at line 10942 @> WARNING failed to parse beta-factor at line 10943 @> WARNING failed to parse beta-factor at line 10944 @> WARNING failed to parse beta-factor at line 10945 @> WARNING failed to parse beta-factor at line 10946 @> WARNING failed to parse beta-factor at line 10947 @> WARNING failed to parse beta-factor at line 10948 @> WARNING failed to parse beta-factor at line 10949 @> WARNING failed to parse beta-factor at line 10950 @> WARNING failed to parse beta-factor at line 10951 @> WARNING failed to parse beta-factor at line 10952 @> WARNING failed to parse beta-factor at line 10953 @> WARNING failed to parse beta-factor at line 10954 @> WARNING failed to parse beta-factor at line 10955 @> WARNING failed to parse beta-factor at line 10956 @> WARNING failed to parse beta-factor at line 10957 @> WARNING failed to parse beta-factor at line 10958 @> WARNING failed to parse beta-factor at line 10959 @> WARNING failed to parse beta-factor at line 10960 @> WARNING failed to parse beta-factor at line 10961 @> WARNING failed to parse beta-factor at line 10962 @> WARNING failed to parse beta-factor at line 10963 @> WARNING failed to parse beta-factor at line 10964 @> WARNING failed to parse beta-factor at line 10965 @> WARNING failed to parse beta-factor at line 10966 @> WARNING failed to parse beta-factor at line 10967 @> WARNING failed to parse beta-factor at line 10968 @> WARNING failed to parse beta-factor at line 10969 @> WARNING failed to parse beta-factor at line 10970 @> WARNING failed to parse beta-factor at line 10971 @> WARNING failed to parse beta-factor at line 10972 @> WARNING failed to parse beta-factor at line 10973 @> WARNING failed to parse beta-factor at line 10974 @> WARNING failed to parse beta-factor at line 10975 @> WARNING failed to parse beta-factor at line 10976 @> WARNING failed to parse beta-factor at line 10977 @> WARNING failed to parse beta-factor at line 10978 @> WARNING failed to parse beta-factor at line 10979 @> WARNING failed to parse beta-factor at line 10980 @> WARNING failed to parse beta-factor at line 10981 @> WARNING failed to parse beta-factor at line 10982 @> WARNING failed to parse beta-factor at line 10983 @> WARNING failed to parse beta-factor at line 10984 @> WARNING failed to parse beta-factor at line 10985 @> WARNING failed to parse beta-factor at line 10986 @> WARNING failed to parse beta-factor at line 10987 @> WARNING failed to parse beta-factor at line 10988 @> WARNING failed to parse beta-factor at line 10989 @> WARNING failed to parse beta-factor at line 10990 @> WARNING failed to parse beta-factor at line 10991 @> WARNING failed to parse beta-factor at line 10992 @> WARNING failed to parse beta-factor at line 10993 @> WARNING failed to parse beta-factor at line 10994 @> WARNING failed to parse beta-factor at line 10995 @> WARNING failed to parse beta-factor at line 10996 @> WARNING failed to parse beta-factor at line 10997 @> WARNING failed to parse beta-factor at line 10998 @> WARNING failed to parse beta-factor at line 10999 @> WARNING failed to parse beta-factor at line 11000 @> WARNING failed to parse beta-factor at line 11001 @> WARNING failed to parse beta-factor at line 11002 @> WARNING failed to parse beta-factor at line 11003 @> WARNING failed to parse beta-factor at line 11004 @> WARNING failed to parse beta-factor at line 11005 @> WARNING failed to parse beta-factor at line 11006 @> WARNING failed to parse beta-factor at line 11007 @> WARNING failed to parse beta-factor at line 11008 @> WARNING failed to parse beta-factor at line 11009 @> WARNING failed to parse beta-factor at line 11010 @> WARNING failed to parse beta-factor at line 11011 @> WARNING failed to parse beta-factor at line 11012 @> WARNING failed to parse beta-factor at line 11013 @> WARNING failed to parse beta-factor at line 11014 @> WARNING failed to parse beta-factor at line 11015 @> WARNING failed to parse beta-factor at line 11016 @> WARNING failed to parse beta-factor at line 11017 @> WARNING failed to parse beta-factor at line 11018 @> WARNING failed to parse beta-factor at line 11019 @> WARNING failed to parse beta-factor at line 11020 @> WARNING failed to parse beta-factor at line 11021 @> WARNING failed to parse beta-factor at line 11022 @> WARNING failed to parse beta-factor at line 11023 @> WARNING failed to parse beta-factor at line 11024 @> WARNING failed to parse beta-factor at line 11025 @> WARNING failed to parse beta-factor at line 11026 @> WARNING failed to parse beta-factor at line 11027 @> WARNING failed to parse beta-factor at line 11028 @> WARNING failed to parse beta-factor at line 11029 @> WARNING failed to parse beta-factor at line 11030 @> WARNING failed to parse beta-factor at line 11031 @> WARNING failed to parse beta-factor at line 11032 @> WARNING failed to parse beta-factor at line 11033 @> WARNING failed to parse beta-factor at line 11034 @> WARNING failed to parse beta-factor at line 11035 @> WARNING failed to parse beta-factor at line 11036 @> WARNING failed to parse beta-factor at line 11037 @> WARNING failed to parse beta-factor at line 11038 @> WARNING failed to parse beta-factor at line 11039 @> WARNING failed to parse beta-factor at line 11040 @> WARNING failed to parse beta-factor at line 11041 @> WARNING failed to parse beta-factor at line 11042 @> WARNING failed to parse beta-factor at line 11043 @> WARNING failed to parse beta-factor at line 11044 @> WARNING failed to parse beta-factor at line 11045 @> WARNING failed to parse beta-factor at line 11046 @> WARNING failed to parse beta-factor at line 11047 @> WARNING failed to parse beta-factor at line 11048 @> WARNING failed to parse beta-factor at line 11049 @> WARNING failed to parse beta-factor at line 11050 @> WARNING failed to parse beta-factor at line 11051 @> WARNING failed to parse beta-factor at line 11052 @> WARNING failed to parse beta-factor at line 11053 @> WARNING failed to parse beta-factor at line 11054 @> WARNING failed to parse beta-factor at line 11055 @> WARNING failed to parse beta-factor at line 11056 @> WARNING failed to parse beta-factor at line 11057 @> WARNING failed to parse beta-factor at line 11058 @> WARNING failed to parse beta-factor at line 11059 @> WARNING failed to parse beta-factor at line 11060 @> WARNING failed to parse beta-factor at line 11061 @> WARNING failed to parse beta-factor at line 11062 @> WARNING failed to parse beta-factor at line 11063 @> WARNING failed to parse beta-factor at line 11064 @> WARNING failed to parse beta-factor at line 11065 @> WARNING failed to parse beta-factor at line 11066 @> WARNING failed to parse beta-factor at line 11067 @> WARNING failed to parse beta-factor at line 11068 @> WARNING failed to parse beta-factor at line 11069 @> WARNING failed to parse beta-factor at line 11070 @> WARNING failed to parse beta-factor at line 11071 @> WARNING failed to parse beta-factor at line 11072 @> WARNING failed to parse beta-factor at line 11073 @> WARNING failed to parse beta-factor at line 11074 @> WARNING failed to parse beta-factor at line 11075 @> WARNING failed to parse beta-factor at line 11076 @> WARNING failed to parse beta-factor at line 11077 @> WARNING failed to parse beta-factor at line 11078 @> WARNING failed to parse beta-factor at line 11079 @> WARNING failed to parse beta-factor at line 11080 @> WARNING failed to parse beta-factor at line 11081 @> WARNING failed to parse beta-factor at line 11082 @> WARNING failed to parse beta-factor at line 11083 @> WARNING failed to parse beta-factor at line 11084 @> WARNING failed to parse beta-factor at line 11085 @> WARNING failed to parse beta-factor at line 11086 @> WARNING failed to parse beta-factor at line 11087 @> WARNING failed to parse beta-factor at line 11088 @> WARNING failed to parse beta-factor at line 11089 @> WARNING failed to parse beta-factor at line 11090 @> WARNING failed to parse beta-factor at line 11091 @> WARNING failed to parse beta-factor at line 11092 @> WARNING failed to parse beta-factor at line 11093 @> WARNING failed to parse beta-factor at line 11094 @> WARNING failed to parse beta-factor at line 11095 @> WARNING failed to parse beta-factor at line 11096 @> WARNING failed to parse beta-factor at line 11097 @> WARNING failed to parse beta-factor at line 11098 @> WARNING failed to parse beta-factor at line 11099 @> WARNING failed to parse beta-factor at line 11100 @> WARNING failed to parse beta-factor at line 11101 @> WARNING failed to parse beta-factor at line 11102 @> WARNING failed to parse beta-factor at line 11103 @> WARNING failed to parse beta-factor at line 11104 @> WARNING failed to parse beta-factor at line 11105 @> WARNING failed to parse beta-factor at line 11106 @> WARNING failed to parse beta-factor at line 11107 @> WARNING failed to parse beta-factor at line 11108 @> WARNING failed to parse beta-factor at line 11109 @> WARNING failed to parse beta-factor at line 11110 @> WARNING failed to parse beta-factor at line 11111 @> WARNING failed to parse beta-factor at line 11112 @> WARNING failed to parse beta-factor at line 11113 @> WARNING failed to parse beta-factor at line 11114 @> WARNING failed to parse beta-factor at line 11115 @> WARNING failed to parse beta-factor at line 11116 @> WARNING failed to parse beta-factor at line 11117 @> WARNING failed to parse beta-factor at line 11118 @> WARNING failed to parse beta-factor at line 11119 @> WARNING failed to parse beta-factor at line 11120 @> WARNING failed to parse beta-factor at line 11121 @> WARNING failed to parse beta-factor at line 11122 @> WARNING failed to parse beta-factor at line 11123 @> WARNING failed to parse beta-factor at line 11124 @> WARNING failed to parse beta-factor at line 11125 @> WARNING failed to parse beta-factor at line 11126 @> WARNING failed to parse beta-factor at line 11127 @> WARNING failed to parse beta-factor at line 11128 @> WARNING failed to parse beta-factor at line 11129 @> WARNING failed to parse beta-factor at line 11130 @> WARNING failed to parse beta-factor at line 11131 @> WARNING failed to parse beta-factor at line 11132 @> WARNING failed to parse beta-factor at line 11133 @> WARNING failed to parse beta-factor at line 11134 @> WARNING failed to parse beta-factor at line 11135 @> WARNING failed to parse beta-factor at line 11136 @> WARNING failed to parse beta-factor at line 11137 @> WARNING failed to parse beta-factor at line 11138 @> WARNING failed to parse beta-factor at line 11139 @> WARNING failed to parse beta-factor at line 11140 @> WARNING failed to parse beta-factor at line 11141 @> WARNING failed to parse beta-factor at line 11142 @> WARNING failed to parse beta-factor at line 11143 @> WARNING failed to parse beta-factor at line 11144 @> WARNING failed to parse beta-factor at line 11145 @> WARNING failed to parse beta-factor at line 11146 @> WARNING failed to parse beta-factor at line 11147 @> WARNING failed to parse beta-factor at line 11148 @> WARNING failed to parse beta-factor at line 11149 @> WARNING failed to parse beta-factor at line 11150 @> WARNING failed to parse beta-factor at line 11151 @> WARNING failed to parse beta-factor at line 11152 @> WARNING failed to parse beta-factor at line 11153 @> WARNING failed to parse beta-factor at line 11154 @> WARNING failed to parse beta-factor at line 11155 @> WARNING failed to parse beta-factor at line 11156 @> WARNING failed to parse beta-factor at line 11157 @> WARNING failed to parse beta-factor at line 11158 @> WARNING failed to parse beta-factor at line 11159 @> WARNING failed to parse beta-factor at line 11160 @> WARNING failed to parse beta-factor at line 11161 @> WARNING failed to parse beta-factor at line 11162 @> WARNING failed to parse beta-factor at line 11163 @> WARNING failed to parse beta-factor at line 11164 @> WARNING failed to parse beta-factor at line 11165 @> WARNING failed to parse beta-factor at line 11166 @> WARNING failed to parse beta-factor at line 11167 @> WARNING failed to parse beta-factor at line 11168 @> WARNING failed to parse beta-factor at line 11169 @> WARNING failed to parse beta-factor at line 11170 @> WARNING failed to parse beta-factor at line 11171 @> WARNING failed to parse beta-factor at line 11172 @> WARNING failed to parse beta-factor at line 11173 @> WARNING failed to parse beta-factor at line 11174 @> WARNING failed to parse beta-factor at line 11175 @> WARNING failed to parse beta-factor at line 11176 @> WARNING failed to parse beta-factor at line 11177 @> WARNING failed to parse beta-factor at line 11178 @> WARNING failed to parse beta-factor at line 11179 @> WARNING failed to parse beta-factor at line 11180 @> WARNING failed to parse beta-factor at line 11181 @> WARNING failed to parse beta-factor at line 11182 @> WARNING failed to parse beta-factor at line 11183 @> WARNING failed to parse beta-factor at line 11184 @> WARNING failed to parse beta-factor at line 11185 @> WARNING failed to parse beta-factor at line 11186 @> WARNING failed to parse beta-factor at line 11187 @> WARNING failed to parse beta-factor at line 11188 @> WARNING failed to parse beta-factor at line 11189 @> WARNING failed to parse beta-factor at line 11190 @> WARNING failed to parse beta-factor at line 11191 @> WARNING failed to parse beta-factor at line 11192 @> WARNING failed to parse beta-factor at line 11193 @> WARNING failed to parse beta-factor at line 11194 @> WARNING failed to parse beta-factor at line 11195 @> WARNING failed to parse beta-factor at line 11196 @> WARNING failed to parse beta-factor at line 11197 @> WARNING failed to parse beta-factor at line 11198 @> WARNING failed to parse beta-factor at line 11199 @> WARNING failed to parse beta-factor at line 11200 @> WARNING failed to parse beta-factor at line 11201 @> WARNING failed to parse beta-factor at line 11202 @> WARNING failed to parse beta-factor at line 11203 @> WARNING failed to parse beta-factor at line 11204 @> WARNING failed to parse beta-factor at line 11205 @> WARNING failed to parse beta-factor at line 11206 @> WARNING failed to parse beta-factor at line 11207 @> WARNING failed to parse beta-factor at line 11208 @> WARNING failed to parse beta-factor at line 11209 @> WARNING failed to parse beta-factor at line 11210 @> WARNING failed to parse beta-factor at line 11211 @> WARNING failed to parse beta-factor at line 11212 @> WARNING failed to parse beta-factor at line 11213 @> WARNING failed to parse beta-factor at line 11214 @> WARNING failed to parse beta-factor at line 11215 @> WARNING failed to parse beta-factor at line 11216 @> WARNING failed to parse beta-factor at line 11217 @> WARNING failed to parse beta-factor at line 11218 @> WARNING failed to parse beta-factor at line 11219 @> WARNING failed to parse beta-factor at line 11220 @> WARNING failed to parse beta-factor at line 11221 @> WARNING failed to parse beta-factor at line 11222 @> WARNING failed to parse beta-factor at line 11223 @> WARNING failed to parse beta-factor at line 11224 @> WARNING failed to parse beta-factor at line 11225 @> WARNING failed to parse beta-factor at line 11226 @> WARNING failed to parse beta-factor at line 11227 @> WARNING failed to parse beta-factor at line 11228 @> WARNING failed to parse beta-factor at line 11229 @> WARNING failed to parse beta-factor at line 11230 @> WARNING failed to parse beta-factor at line 11231 @> WARNING failed to parse beta-factor at line 11232 @> WARNING failed to parse beta-factor at line 11233 @> WARNING failed to parse beta-factor at line 11234 @> WARNING failed to parse beta-factor at line 11235 @> WARNING failed to parse beta-factor at line 11236 @> WARNING failed to parse beta-factor at line 11237 @> WARNING failed to parse beta-factor at line 11238 @> WARNING failed to parse beta-factor at line 11239 @> WARNING failed to parse beta-factor at line 11240 @> WARNING failed to parse beta-factor at line 11241 @> WARNING failed to parse beta-factor at line 11242 @> WARNING failed to parse beta-factor at line 11243 @> WARNING failed to parse beta-factor at line 11244 @> WARNING failed to parse beta-factor at line 11245 @> WARNING failed to parse beta-factor at line 11246 @> WARNING failed to parse beta-factor at line 11247 @> WARNING failed to parse beta-factor at line 11248 @> WARNING failed to parse beta-factor at line 11249 @> WARNING failed to parse beta-factor at line 11250 @> WARNING failed to parse beta-factor at line 11251 @> WARNING failed to parse beta-factor at line 11252 @> WARNING failed to parse beta-factor at line 11253 @> WARNING failed to parse beta-factor at line 11254 @> WARNING failed to parse beta-factor at line 11255 @> WARNING failed to parse beta-factor at line 11256 @> WARNING failed to parse beta-factor at line 11257 @> WARNING failed to parse beta-factor at line 11258 @> WARNING failed to parse beta-factor at line 11259 @> WARNING failed to parse beta-factor at line 11260 @> WARNING failed to parse beta-factor at line 11261 @> WARNING failed to parse beta-factor at line 11262 @> WARNING failed to parse beta-factor at line 11263 @> WARNING failed to parse beta-factor at line 11264 @> WARNING failed to parse beta-factor at line 11265 @> WARNING failed to parse beta-factor at line 11266 @> WARNING failed to parse beta-factor at line 11267 @> WARNING failed to parse beta-factor at line 11268 @> WARNING failed to parse beta-factor at line 11269 @> WARNING failed to parse beta-factor at line 11270 @> WARNING failed to parse beta-factor at line 11271 @> WARNING failed to parse beta-factor at line 11272 @> WARNING failed to parse beta-factor at line 11273 @> WARNING failed to parse beta-factor at line 11274 @> WARNING failed to parse beta-factor at line 11275 @> WARNING failed to parse beta-factor at line 11276 @> WARNING failed to parse beta-factor at line 11277 @> WARNING failed to parse beta-factor at line 11278 @> WARNING failed to parse beta-factor at line 11279 @> WARNING failed to parse beta-factor at line 11280 @> WARNING failed to parse beta-factor at line 11281 @> WARNING failed to parse beta-factor at line 11282 @> WARNING failed to parse beta-factor at line 11283 @> WARNING failed to parse beta-factor at line 11284 @> WARNING failed to parse beta-factor at line 11285 @> WARNING failed to parse beta-factor at line 11286 @> WARNING failed to parse beta-factor at line 11287 @> WARNING failed to parse beta-factor at line 11288 @> WARNING failed to parse beta-factor at line 11289 @> WARNING failed to parse beta-factor at line 11290 @> WARNING failed to parse beta-factor at line 11291 @> WARNING failed to parse beta-factor at line 11292 @> WARNING failed to parse beta-factor at line 11293 @> WARNING failed to parse beta-factor at line 11294 @> WARNING failed to parse beta-factor at line 11295 @> WARNING failed to parse beta-factor at line 11296 @> WARNING failed to parse beta-factor at line 11297 @> WARNING failed to parse beta-factor at line 11298 @> WARNING failed to parse beta-factor at line 11299 @> WARNING failed to parse beta-factor at line 11300 @> WARNING failed to parse beta-factor at line 11301 @> WARNING failed to parse beta-factor at line 11302 @> WARNING failed to parse beta-factor at line 11303 @> WARNING failed to parse beta-factor at line 11304 @> WARNING failed to parse beta-factor at line 11305 @> WARNING failed to parse beta-factor at line 11306 @> WARNING failed to parse beta-factor at line 11307 @> WARNING failed to parse beta-factor at line 11308 @> WARNING failed to parse beta-factor at line 11309 @> WARNING failed to parse beta-factor at line 11310 @> WARNING failed to parse beta-factor at line 11311 @> WARNING failed to parse beta-factor at line 11312 @> WARNING failed to parse beta-factor at line 11313 @> WARNING failed to parse beta-factor at line 11314 @> WARNING failed to parse beta-factor at line 11315 @> WARNING failed to parse beta-factor at line 11316 @> WARNING failed to parse beta-factor at line 11317 @> WARNING failed to parse beta-factor at line 11318 @> WARNING failed to parse beta-factor at line 11319 @> WARNING failed to parse beta-factor at line 11320 @> WARNING failed to parse beta-factor at line 11321 @> WARNING failed to parse beta-factor at line 11322 @> WARNING failed to parse beta-factor at line 11323 @> WARNING failed to parse beta-factor at line 11324 @> WARNING failed to parse beta-factor at line 11325 @> WARNING failed to parse beta-factor at line 11326 @> WARNING failed to parse beta-factor at line 11327 @> WARNING failed to parse beta-factor at line 11328 @> WARNING failed to parse beta-factor at line 11329 @> WARNING failed to parse beta-factor at line 11330 @> WARNING failed to parse beta-factor at line 11331 @> WARNING failed to parse beta-factor at line 11332 @> WARNING failed to parse beta-factor at line 11333 @> WARNING failed to parse beta-factor at line 11334 @> WARNING failed to parse beta-factor at line 11335 @> WARNING failed to parse beta-factor at line 11336 @> WARNING failed to parse beta-factor at line 11337 @> WARNING failed to parse beta-factor at line 11338 @> WARNING failed to parse beta-factor at line 11339 @> WARNING failed to parse beta-factor at line 11340 @> WARNING failed to parse beta-factor at line 11341 @> WARNING failed to parse beta-factor at line 11342 @> WARNING failed to parse beta-factor at line 11343 @> WARNING failed to parse beta-factor at line 11344 @> WARNING failed to parse beta-factor at line 11345 @> WARNING failed to parse beta-factor at line 11346 @> WARNING failed to parse beta-factor at line 11347 @> WARNING failed to parse beta-factor at line 11348 @> WARNING failed to parse beta-factor at line 11349 @> WARNING failed to parse beta-factor at line 11350 @> WARNING failed to parse beta-factor at line 11351 @> WARNING failed to parse beta-factor at line 11352 @> WARNING failed to parse beta-factor at line 11353 @> WARNING failed to parse beta-factor at line 11354 @> WARNING failed to parse beta-factor at line 11355 @> WARNING failed to parse beta-factor at line 11356 @> WARNING failed to parse beta-factor at line 11357 @> WARNING failed to parse beta-factor at line 11358 @> WARNING failed to parse beta-factor at line 11359 @> WARNING failed to parse beta-factor at line 11360 @> WARNING failed to parse beta-factor at line 11361 @> WARNING failed to parse beta-factor at line 11362 @> WARNING failed to parse beta-factor at line 11363 @> WARNING failed to parse beta-factor at line 11364 @> WARNING failed to parse beta-factor at line 11365 @> WARNING failed to parse beta-factor at line 11366 @> WARNING failed to parse beta-factor at line 11367 @> WARNING failed to parse beta-factor at line 11368 @> WARNING failed to parse beta-factor at line 11369 @> WARNING failed to parse beta-factor at line 11370 @> WARNING failed to parse beta-factor at line 11371 @> WARNING failed to parse beta-factor at line 11372 @> WARNING failed to parse beta-factor at line 11373 @> WARNING failed to parse beta-factor at line 11374 @> WARNING failed to parse beta-factor at line 11375 @> WARNING failed to parse beta-factor at line 11376 @> WARNING failed to parse beta-factor at line 11377 @> WARNING failed to parse beta-factor at line 11378 @> WARNING failed to parse beta-factor at line 11379 @> WARNING failed to parse beta-factor at line 11380 @> WARNING failed to parse beta-factor at line 11381 @> WARNING failed to parse beta-factor at line 11382 @> WARNING failed to parse beta-factor at line 11383 @> WARNING failed to parse beta-factor at line 11384 @> WARNING failed to parse beta-factor at line 11385 @> WARNING failed to parse beta-factor at line 11386 @> WARNING failed to parse beta-factor at line 11387 @> WARNING failed to parse beta-factor at line 11388 @> WARNING failed to parse beta-factor at line 11389 @> WARNING failed to parse beta-factor at line 11390 @> WARNING failed to parse beta-factor at line 11391 @> WARNING failed to parse beta-factor at line 11392 @> WARNING failed to parse beta-factor at line 11393 @> WARNING failed to parse beta-factor at line 11394 @> WARNING failed to parse beta-factor at line 11395 @> WARNING failed to parse beta-factor at line 11396 @> WARNING failed to parse beta-factor at line 11397 @> WARNING failed to parse beta-factor at line 11398 @> WARNING failed to parse beta-factor at line 11399 @> WARNING failed to parse beta-factor at line 11400 @> WARNING failed to parse beta-factor at line 11401 @> WARNING failed to parse beta-factor at line 11402 @> WARNING failed to parse beta-factor at line 11403 @> WARNING failed to parse beta-factor at line 11404 @> WARNING failed to parse beta-factor at line 11405 @> WARNING failed to parse beta-factor at line 11406 @> WARNING failed to parse beta-factor at line 11407 @> WARNING failed to parse beta-factor at line 11408 @> WARNING failed to parse beta-factor at line 11409 @> WARNING failed to parse beta-factor at line 11410 @> WARNING failed to parse beta-factor at line 11411 @> WARNING failed to parse beta-factor at line 11412 @> WARNING failed to parse beta-factor at line 11413 @> WARNING failed to parse beta-factor at line 11414 @> WARNING failed to parse beta-factor at line 11415 @> WARNING failed to parse beta-factor at line 11416 @> WARNING failed to parse beta-factor at line 11417 @> WARNING failed to parse beta-factor at line 11418 @> WARNING failed to parse beta-factor at line 11419 @> WARNING failed to parse beta-factor at line 11420 @> WARNING failed to parse beta-factor at line 11421 @> WARNING failed to parse beta-factor at line 11422 @> WARNING failed to parse beta-factor at line 11423 @> WARNING failed to parse beta-factor at line 11424 @> WARNING failed to parse beta-factor at line 11425 @> WARNING failed to parse beta-factor at line 11426 @> WARNING failed to parse beta-factor at line 11427 @> WARNING failed to parse beta-factor at line 11428 @> WARNING failed to parse beta-factor at line 11429 @> WARNING failed to parse beta-factor at line 11430 @> WARNING failed to parse beta-factor at line 11431 @> WARNING failed to parse beta-factor at line 11432 @> WARNING failed to parse beta-factor at line 11433 @> WARNING failed to parse beta-factor at line 11434 @> WARNING failed to parse beta-factor at line 11435 @> WARNING failed to parse beta-factor at line 11436 @> WARNING failed to parse beta-factor at line 11437 @> WARNING failed to parse beta-factor at line 11438 @> WARNING failed to parse beta-factor at line 11439 @> WARNING failed to parse beta-factor at line 11440 @> WARNING failed to parse beta-factor at line 11441 @> WARNING failed to parse beta-factor at line 11442 @> WARNING failed to parse beta-factor at line 11443 @> WARNING failed to parse beta-factor at line 11444 @> WARNING failed to parse beta-factor at line 11445 @> WARNING failed to parse beta-factor at line 11446 @> WARNING failed to parse beta-factor at line 11447 @> WARNING failed to parse beta-factor at line 11448 @> WARNING failed to parse beta-factor at line 11449 @> WARNING failed to parse beta-factor at line 11450 @> WARNING failed to parse beta-factor at line 11451 @> WARNING failed to parse beta-factor at line 11452 @> WARNING failed to parse beta-factor at line 11453 @> WARNING failed to parse beta-factor at line 11454 @> WARNING failed to parse beta-factor at line 11455 @> WARNING failed to parse beta-factor at line 11456 @> WARNING failed to parse beta-factor at line 11457 @> WARNING failed to parse beta-factor at line 11458 @> WARNING failed to parse beta-factor at line 11459 @> WARNING failed to parse beta-factor at line 11460 @> WARNING failed to parse beta-factor at line 11461 @> WARNING failed to parse beta-factor at line 11462 @> WARNING failed to parse beta-factor at line 11463 @> WARNING failed to parse beta-factor at line 11464 @> WARNING failed to parse beta-factor at line 11465 @> WARNING failed to parse beta-factor at line 11466 @> WARNING failed to parse beta-factor at line 11467 @> WARNING failed to parse beta-factor at line 11468 @> WARNING failed to parse beta-factor at line 11469 @> WARNING failed to parse beta-factor at line 11470 @> WARNING failed to parse beta-factor at line 11471 @> WARNING failed to parse beta-factor at line 11472 @> WARNING failed to parse beta-factor at line 11473 @> WARNING failed to parse beta-factor at line 11474 @> WARNING failed to parse beta-factor at line 11475 @> WARNING failed to parse beta-factor at line 11476 @> WARNING failed to parse beta-factor at line 11477 @> WARNING failed to parse beta-factor at line 11478 @> WARNING failed to parse beta-factor at line 11479 @> WARNING failed to parse beta-factor at line 11480 @> WARNING failed to parse beta-factor at line 11481 @> WARNING failed to parse beta-factor at line 11482 @> WARNING failed to parse beta-factor at line 11483 @> WARNING failed to parse beta-factor at line 11484 @> WARNING failed to parse beta-factor at line 11485 @> WARNING failed to parse beta-factor at line 11486 @> WARNING failed to parse beta-factor at line 11487 @> WARNING failed to parse beta-factor at line 11488 @> WARNING failed to parse beta-factor at line 11489 @> WARNING failed to parse beta-factor at line 11490 @> WARNING failed to parse beta-factor at line 11491 @> WARNING failed to parse beta-factor at line 11492 @> WARNING failed to parse beta-factor at line 11493 @> WARNING failed to parse beta-factor at line 11494 @> WARNING failed to parse beta-factor at line 11495 @> WARNING failed to parse beta-factor at line 11496 @> WARNING failed to parse beta-factor at line 11497 @> WARNING failed to parse beta-factor at line 11498 @> WARNING failed to parse beta-factor at line 11499 @> WARNING failed to parse beta-factor at line 11500 @> WARNING failed to parse beta-factor at line 11501 @> WARNING failed to parse beta-factor at line 11502 @> WARNING failed to parse beta-factor at line 11503 @> WARNING failed to parse beta-factor at line 11504 @> WARNING failed to parse beta-factor at line 11505 @> WARNING failed to parse beta-factor at line 11506 @> WARNING failed to parse beta-factor at line 11507 @> WARNING failed to parse beta-factor at line 11508 @> WARNING failed to parse beta-factor at line 11509 @> WARNING failed to parse beta-factor at line 11510 @> WARNING failed to parse beta-factor at line 11511 @> WARNING failed to parse beta-factor at line 11512 @> WARNING failed to parse beta-factor at line 11513 @> WARNING failed to parse beta-factor at line 11514 @> WARNING failed to parse beta-factor at line 11515 @> WARNING failed to parse beta-factor at line 11516 @> WARNING failed to parse beta-factor at line 11517 @> WARNING failed to parse beta-factor at line 11518 @> WARNING failed to parse beta-factor at line 11519 @> WARNING failed to parse beta-factor at line 11520 @> WARNING failed to parse beta-factor at line 11521 @> WARNING failed to parse beta-factor at line 11522 @> WARNING failed to parse beta-factor at line 11523 @> WARNING failed to parse beta-factor at line 11524 @> WARNING failed to parse beta-factor at line 11525 @> WARNING failed to parse beta-factor at line 11526 @> WARNING failed to parse beta-factor at line 11527 @> WARNING failed to parse beta-factor at line 11528 @> WARNING failed to parse beta-factor at line 11529 @> WARNING failed to parse beta-factor at line 11530 @> WARNING failed to parse beta-factor at line 11531 @> WARNING failed to parse beta-factor at line 11532 @> WARNING failed to parse beta-factor at line 11533 @> WARNING failed to parse beta-factor at line 11534 @> WARNING failed to parse beta-factor at line 11535 @> WARNING failed to parse beta-factor at line 11536 @> WARNING failed to parse beta-factor at line 11537 @> WARNING failed to parse beta-factor at line 11538 @> WARNING failed to parse beta-factor at line 11539 @> WARNING failed to parse beta-factor at line 11540 @> WARNING failed to parse beta-factor at line 11541 @> WARNING failed to parse beta-factor at line 11542 @> WARNING failed to parse beta-factor at line 11543 @> WARNING failed to parse beta-factor at line 11544 @> WARNING failed to parse beta-factor at line 11545 @> WARNING failed to parse beta-factor at line 11546 @> WARNING failed to parse beta-factor at line 11547 @> WARNING failed to parse beta-factor at line 11548 @> WARNING failed to parse beta-factor at line 11549 @> WARNING failed to parse beta-factor at line 11550 @> WARNING failed to parse beta-factor at line 11551 @> WARNING failed to parse beta-factor at line 11552 @> WARNING failed to parse beta-factor at line 11553 @> WARNING failed to parse beta-factor at line 11554 @> WARNING failed to parse beta-factor at line 11555 @> WARNING failed to parse beta-factor at line 11556 @> WARNING failed to parse beta-factor at line 11557 @> WARNING failed to parse beta-factor at line 11558 @> WARNING failed to parse beta-factor at line 11559 @> WARNING failed to parse beta-factor at line 11560 @> WARNING failed to parse beta-factor at line 11561 @> WARNING failed to parse beta-factor at line 11562 @> WARNING failed to parse beta-factor at line 11563 @> WARNING failed to parse beta-factor at line 11564 @> WARNING failed to parse beta-factor at line 11565 @> WARNING failed to parse beta-factor at line 11566 @> WARNING failed to parse beta-factor at line 11567 @> WARNING failed to parse beta-factor at line 11568 @> WARNING failed to parse beta-factor at line 11569 @> WARNING failed to parse beta-factor at line 11570 @> WARNING failed to parse beta-factor at line 11571 @> WARNING failed to parse beta-factor at line 11572 @> WARNING failed to parse beta-factor at line 11573 @> WARNING failed to parse beta-factor at line 11574 @> WARNING failed to parse beta-factor at line 11575 @> WARNING failed to parse beta-factor at line 11576 @> WARNING failed to parse beta-factor at line 11577 @> WARNING failed to parse beta-factor at line 11578 @> WARNING failed to parse beta-factor at line 11579 @> WARNING failed to parse beta-factor at line 11580 @> WARNING failed to parse beta-factor at line 11581 @> WARNING failed to parse beta-factor at line 11582 @> WARNING failed to parse beta-factor at line 11583 @> WARNING failed to parse beta-factor at line 11584 @> WARNING failed to parse beta-factor at line 11585 @> WARNING failed to parse beta-factor at line 11586 @> WARNING failed to parse beta-factor at line 11587 @> WARNING failed to parse beta-factor at line 11588 @> WARNING failed to parse beta-factor at line 11589 @> WARNING failed to parse beta-factor at line 11590 @> WARNING failed to parse beta-factor at line 11591 @> WARNING failed to parse beta-factor at line 11592 @> WARNING failed to parse beta-factor at line 11593 @> WARNING failed to parse beta-factor at line 11594 @> WARNING failed to parse beta-factor at line 11595 @> WARNING failed to parse beta-factor at line 11596 @> WARNING failed to parse beta-factor at line 11597 @> WARNING failed to parse beta-factor at line 11598 @> WARNING failed to parse beta-factor at line 11599 @> WARNING failed to parse beta-factor at line 11600 @> WARNING failed to parse beta-factor at line 11601 @> WARNING failed to parse beta-factor at line 11602 @> WARNING failed to parse beta-factor at line 11603 @> WARNING failed to parse beta-factor at line 11604 @> WARNING failed to parse beta-factor at line 11605 @> WARNING failed to parse beta-factor at line 11606 @> WARNING failed to parse beta-factor at line 11607 @> WARNING failed to parse beta-factor at line 11608 @> WARNING failed to parse beta-factor at line 11609 @> WARNING failed to parse beta-factor at line 11610 @> WARNING failed to parse beta-factor at line 11611 @> WARNING failed to parse beta-factor at line 11612 @> WARNING failed to parse beta-factor at line 11613 @> WARNING failed to parse beta-factor at line 11614 @> WARNING failed to parse beta-factor at line 11615 @> WARNING failed to parse beta-factor at line 11616 @> WARNING failed to parse beta-factor at line 11617 @> WARNING failed to parse beta-factor at line 11618 @> WARNING failed to parse beta-factor at line 11619 @> WARNING failed to parse beta-factor at line 11620 @> WARNING failed to parse beta-factor at line 11621 @> WARNING failed to parse beta-factor at line 11622 @> WARNING failed to parse beta-factor at line 11623 @> WARNING failed to parse beta-factor at line 11624 @> WARNING failed to parse beta-factor at line 11625 @> WARNING failed to parse beta-factor at line 11626 @> WARNING failed to parse beta-factor at line 11627 @> WARNING failed to parse beta-factor at line 11628 @> WARNING failed to parse beta-factor at line 11629 @> WARNING failed to parse beta-factor at line 11630 @> WARNING failed to parse beta-factor at line 11631 @> WARNING failed to parse beta-factor at line 11632 @> WARNING failed to parse beta-factor at line 11633 @> WARNING failed to parse beta-factor at line 11634 @> WARNING failed to parse beta-factor at line 11635 @> WARNING failed to parse beta-factor at line 11636 @> WARNING failed to parse beta-factor at line 11637 @> WARNING failed to parse beta-factor at line 11638 @> WARNING failed to parse beta-factor at line 11639 @> WARNING failed to parse beta-factor at line 11640 @> WARNING failed to parse beta-factor at line 11641 @> WARNING failed to parse beta-factor at line 11642 @> WARNING failed to parse beta-factor at line 11643 @> WARNING failed to parse beta-factor at line 11644 @> WARNING failed to parse beta-factor at line 11645 @> WARNING failed to parse beta-factor at line 11646 @> WARNING failed to parse beta-factor at line 11647 @> WARNING failed to parse beta-factor at line 11648 @> WARNING failed to parse beta-factor at line 11649 @> WARNING failed to parse beta-factor at line 11650 @> WARNING failed to parse beta-factor at line 11651 @> WARNING failed to parse beta-factor at line 11652 @> WARNING failed to parse beta-factor at line 11653 @> WARNING failed to parse beta-factor at line 11654 @> WARNING failed to parse beta-factor at line 11655 @> WARNING failed to parse beta-factor at line 11656 @> WARNING failed to parse beta-factor at line 11657 @> WARNING failed to parse beta-factor at line 11658 @> WARNING failed to parse beta-factor at line 11659 @> WARNING failed to parse beta-factor at line 11660 @> WARNING failed to parse beta-factor at line 11661 @> WARNING failed to parse beta-factor at line 11662 @> WARNING failed to parse beta-factor at line 11663 @> WARNING failed to parse beta-factor at line 11664 @> WARNING failed to parse beta-factor at line 11665 @> WARNING failed to parse beta-factor at line 11666 @> WARNING failed to parse beta-factor at line 11667 @> WARNING failed to parse beta-factor at line 11668 @> WARNING failed to parse beta-factor at line 11669 @> WARNING failed to parse beta-factor at line 11670 @> WARNING failed to parse beta-factor at line 11671 @> WARNING failed to parse beta-factor at line 11672 @> WARNING failed to parse beta-factor at line 11673 @> WARNING failed to parse beta-factor at line 11674 @> WARNING failed to parse beta-factor at line 11675 @> WARNING failed to parse beta-factor at line 11676 @> WARNING failed to parse beta-factor at line 11677 @> WARNING failed to parse beta-factor at line 11678 @> WARNING failed to parse beta-factor at line 11679 @> WARNING failed to parse beta-factor at line 11680 @> WARNING failed to parse beta-factor at line 11681 @> WARNING failed to parse beta-factor at line 11682 @> WARNING failed to parse beta-factor at line 11683 @> WARNING failed to parse beta-factor at line 11684 @> WARNING failed to parse beta-factor at line 11685 @> WARNING failed to parse beta-factor at line 11686 @> WARNING failed to parse beta-factor at line 11687 @> WARNING failed to parse beta-factor at line 11688 @> WARNING failed to parse beta-factor at line 11689 @> WARNING failed to parse beta-factor at line 11690 @> WARNING failed to parse beta-factor at line 11691 @> WARNING failed to parse beta-factor at line 11692 @> WARNING failed to parse beta-factor at line 11693 @> WARNING failed to parse beta-factor at line 11694 @> WARNING failed to parse beta-factor at line 11695 @> WARNING failed to parse beta-factor at line 11696 @> WARNING failed to parse beta-factor at line 11697 @> WARNING failed to parse beta-factor at line 11698 @> WARNING failed to parse beta-factor at line 11699 @> WARNING failed to parse beta-factor at line 11700 @> WARNING failed to parse beta-factor at line 11701 @> WARNING failed to parse beta-factor at line 11702 @> WARNING failed to parse beta-factor at line 11703 @> WARNING failed to parse beta-factor at line 11704 @> WARNING failed to parse beta-factor at line 11705 @> WARNING failed to parse beta-factor at line 11706 @> WARNING failed to parse beta-factor at line 11707 @> WARNING failed to parse beta-factor at line 11708 @> WARNING failed to parse beta-factor at line 11709 @> WARNING failed to parse beta-factor at line 11710 @> WARNING failed to parse beta-factor at line 11711 @> WARNING failed to parse beta-factor at line 11712 @> WARNING failed to parse beta-factor at line 11713 @> WARNING failed to parse beta-factor at line 11714 @> WARNING failed to parse beta-factor at line 11715 @> WARNING failed to parse beta-factor at line 11716 @> WARNING failed to parse beta-factor at line 11717 @> WARNING failed to parse beta-factor at line 11718 @> WARNING failed to parse beta-factor at line 11719 @> WARNING failed to parse beta-factor at line 11720 @> WARNING failed to parse beta-factor at line 11721 @> WARNING failed to parse beta-factor at line 11722 @> WARNING failed to parse beta-factor at line 11723 @> WARNING failed to parse beta-factor at line 11724 @> WARNING failed to parse beta-factor at line 11725 @> WARNING failed to parse beta-factor at line 11726 @> WARNING failed to parse beta-factor at line 11727 @> WARNING failed to parse beta-factor at line 11728 @> WARNING failed to parse beta-factor at line 11729 IOPub message rate exceeded. The notebook server will temporarily stop sending output to the client in order to avoid crashing it. To change this limit, set the config variable `--NotebookApp.iopub_msg_rate_limit`. Current values: NotebookApp.iopub_msg_rate_limit=1000.0 (msgs/sec) NotebookApp.rate_limit_window=3.0 (secs)
There will be warnings saying that ProDy wants to read beta factors, but the coordinates should be read properly. In addition to atoms, the OPM file contains points to indicate the boundaries of the membrane. We will need this information to specify the thickness of the membrane. To obtain this information, we first inspect the chain IDs,
unique(of_all.getChids())
array([' ', 'A', 'B', 'C', 'D'], dtype='<U2')
The membrane nodes have empty chain IDs. Therefore, we can select them by:
membrane = of_all.select('not chain A B C D')
And then obtain the lower and upper bound of the membrane along the z-axis:
membrane.getCoords()[:, 2].max()
13.423
membrane.getCoords()[:, 2].min()
-13.423
ProDy’s exANM method can be used for any system. This method will create a membrane with given highest and lowest coordinate on the Z-axis. The main advantage of this method is that the protein can interact with lipid molecules on the membrane. The elastic network model based on the interaction between aminoacids on protein and the interaction between aminoacids with the lipids on membrane. The addition of physical membrane will avoid the unphysical distortion of the structure. This will not reduce accuracy as in the case of implicit membrane ANM model.
To use the explicit membrane for ANM calculation, we first select the protein part of the structure:
of_ca = of_all.select('protein and name CA and not (chain A and resid 119 to 122) and not (chain C and resid 119 to 123) and not chain D')
And then instantiate an exANM object:
of_ca.numAtoms()
1194
exanm = exANM('2nwl_ex')
Then we build a couple of Hessians using the coordinates of the crystal structures,
exanm.buildHessian(of_ca, h=15., R=120., turbo=True)
@> Membrane was built in 26s. @> layers: [0 1 2 3 4] @> max layer: 4 @> layer: 0 @> layer: 1 @> layer: 2 @> layer: 3 @> layer: 4 @> layer: 4 finished @> layer: 3 finished @> layer: 2 finished @> layer: 1 finished @> layer: 0 finished @> Hessian was built in 96.83s.
Now we calculate the modes and write them to a pair of .nmd files for viewing.
exanm._membrane
<AtomGroup: Membrane (7282 atoms)>
exanm.calcModes()
writeNMD('2nwl_ex.nmd',exanm,of_ca.select('protein and name CA'))
@> 20 modes were calculated in 2.95s.
'2nwl_ex.nmd'