Sampling Functions¶
This module defines functions for generating alternate conformations along normal modes.
-
deformAtoms(atoms, mode, rmsd=None, replace=False, scale=None)[source]¶ Generate a new coordinate set for atoms along the mode. atoms must be a
AtomGroupinstance. New coordinate set will be appended to atoms. If rmsd is provided, mode will be scaled to generate a coordinate set with given RMSD distance to the active coordinate set.
-
sampleModes(modes, atoms=None, n_confs=1000, rmsd=1.0)[source]¶ Returns an ensemble of randomly sampled conformations along given modes. If atoms are provided, sampling will be around its active coordinate set. Otherwise, sampling is around the 0 coordinate set.
Parameters: - modes (
Mode,ModeSet,PCA,ANMorNMA) – modes along which sampling will be performed - atoms (
Atomic) – atoms whose active coordinate set will be used as the initial conformation - n_confs – number of conformations to generate, default is 1000
- rmsd (float) – average RMSD that the conformations will have with respect to the initial conformation, default is 1.0 Å
Returns: For given normal modes
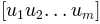 and their eigenvalues
and their eigenvalues
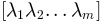 , a new conformation
is sampled using the relation:
, a new conformation
is sampled using the relation: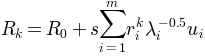
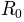 is the active coordinate set of atoms.
is the active coordinate set of atoms.
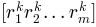 are normally distributed random numbers
generated for conformation
are normally distributed random numbers
generated for conformation 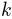 using
using numpy.random.randn().RMSD of the new conformation from
can be calculated as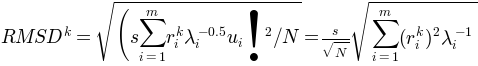
Average
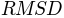 of the generated conformations from the initial conformation is:
of the generated conformations from the initial conformation is: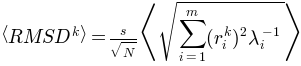
From this relation
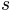 scaling factor obtained using the relation
scaling factor obtained using the relation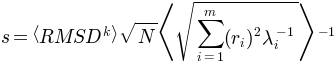
Note that random numbers are generated before conformations are sampled, hence exact value of
is known from this relation to
ensure that the generated ensemble will have user given average rmsd
value.Note that if modes are from a
PCA, variances are used instead of inverse eigenvalues, i.e.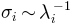 .
.See also
showEllipsoid().- modes (
-
traverseMode(mode, atoms, n_steps=10, rmsd=1.5, **kwargs)[source]¶ Generates a trajectory along a given mode, which can be used to animate fluctuations in an external program.
Parameters: - mode (
Mode) – mode along which a trajectory will be generated - atoms (
Atomic) – atoms whose active coordinate set will be used as the initial conformation - n_steps (int) – number of steps to take along each direction,
for example, for
n_steps=10, 20 conformations will be generated along the first mode, default is 10. - rmsd (float) – maximum RMSD that the conformations will have with respect to the initial conformation, default is 1.5 Å
- pos (bool) – whether to include steps in the positive mode direction, default is True
- neg – whether to include steps in the negative mode direction, default is True
- reverse (bool) – whether to reverse the direction default is False
Returns: For given normal mode
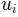 , its eigenvalue
, its eigenvalue
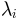 , number of steps
, number of steps 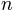 , and maximum
conformations
, and maximum
conformations 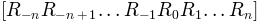 are
generated. is the active coordinate set of atoms.
are
generated. is the active coordinate set of atoms.
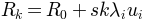 , where is found using
, where is found using
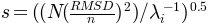 , where
, where
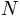 is the number of atoms.
is the number of atoms.- mode (