Analysis Functions¶
This module defines MSA analysis functions.
-
calcShannonEntropy(msa, ambiguity=True, omitgaps=True, **kwargs)[source]¶ Returns Shannon entropy array calculated for msa, which may be an
MSAinstance or a 2D Numpy character array. Implementation is case insensitive and handles ambiguous amino acids as follows:- B (Asx) count is allocated to D (Asp) and N (Asn)
- Z (Glx) count is allocated to E (Glu) and Q (Gln)
- J (Xle) count is allocated to I (Ile) and L (Leu)
- X (Xaa) count is allocated to the twenty standard amino acids
Selenocysteine (U, Sec) and pyrrolysine (O, Pyl) are considered as distinct amino acids. When ambiguity is set False, all alphabet characters as considered as distinct types.
All non-alphabet characters are considered as gaps, and they are handled in two ways:
- non-existent, the probability of observing amino acids in a given column is adjusted, by default
- as a distinct character with its own probability, when omitgaps is False
-
buildMutinfoMatrix(msa, ambiguity=True, turbo=True, **kwargs)[source]¶ Returns mutual information matrix calculated for msa, which may be an
MSAinstance or a 2D Numpy character array. Implementation is case insensitive and handles ambiguous amino acids as follows:- B (Asx) count is allocated to D (Asp) and N (Asn)
- Z (Glx) count is allocated to E (Glu) and Q (Gln)
- J (Xle) count is allocated to I (Ile) and L (Leu)
- X (Xaa) count is allocated to the twenty standard amino acids
- Joint probability of observing a pair of ambiguous amino acids is allocated to all potential combinations, e.g. probability of XX is allocated to 400 combinations of standard amino acids, similarly probability of XB is allocated to 40 combinations of D and N with the standard amino acids.
Selenocysteine (U, Sec) and pyrrolysine (O, Pyl) are considered as distinct amino acids. When ambiguity is set False, all alphabet characters as considered as distinct types. All non-alphabet characters are considered as gaps.
Mutual information matrix can be normalized or corrected using
applyMINormalization()andapplyMICorrection()methods, respectively. Normalization by joint entropy can performed using this function with norm option set True.By default, turbo mode, which uses memory as large as the MSA array itself but runs four to five times faster, will be used. If memory allocation fails, the implementation will fall back to slower and memory efficient mode.
-
calcMSAOccupancy(msa, occ='res', count=False)[source]¶ Returns occupancy array calculated for residue positions (default,
'res'or'col'for occ) or sequences ('seq'or'row'for occ) of msa, which may be anMSAinstance or a 2D NumPy character array. By default, occupancy [0-1] will be calculated. If count is True, count of non-gap characters will be returned. Implementation is case insensitive.
-
applyMutinfoCorr(mutinfo, corr='prod')[source]¶ Returns a copy of mutinfo array after average product correction (default) or average sum correction is applied. See [DSD08] for details.
[DSD08] Dunn SD, Wahl LM, Gloor GB. Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction. Bioinformatics 2008 24(3):333-340.
-
applyMutinfoNorm(mutinfo, entropy, norm='sument')[source]¶ Apply one of the normalizations discussed in [MLC05] to mutinfo matrix. norm can be one of the following:
'sument':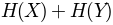 , sum of entropy of columns
, sum of entropy of columns'minent':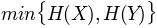 , minimum entropy
, minimum entropy'maxent':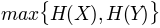 , maximum entropy
, maximum entropy'mincon':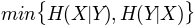 , minimum conditional
, minimum conditionalentropy
'maxcon':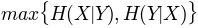 , maximum conditional
, maximum conditionalentropy
where
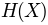 is the entropy of a column, and
is the entropy of a column, and
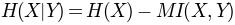 . Normalization with joint entropy, i.e.
. Normalization with joint entropy, i.e.
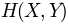 , can be done using
, can be done using buildMutinfoMatrix()norm argument.[MLC05] Martin LC, Gloor GB, Dunn SD, Wahl LM. Using information theory to search for co-evolving residues in proteins. Bioinformatics 2005 21(22):4116-4124.
-
calcRankorder(matrix, zscore=False, **kwargs)[source]¶ Returns indices of elements and corresponding values sorted in descending order, if descend is True (default). Can apply a zscore normalization; by default along axis - 0 such that each column has
mean=0andstd=1. If zcore analysis is used, return value contains the zscores. If matrix is symmetric only lower triangle indices will be returned, with diagonal elements if diag is True (default).
-
filterRankedPairs(pdb, indices, msa_indices, rank_row, rank_col, zscore_sort, num_of_pairs=20, seqDistance=5, resi_range=None, pdbDistance=8, chain1='A', chain2='A')[source]¶ indices and msa_indices are lists output from alignSequenceToMSA
rank_row, rank_col and zscore_sort are the outputs from calcRankorder
Parameters: - num_of_pairs (int) – The number of pairs to be output, if no value is given then all pairs are output. Default is 20
- seqDistance (int) – Remove pairs that are closer than this in the reference sequence Default is 5
- pdbDistance (int) – Remove pairs with Calpha atoms further apart than this in the PDB Default is 8
- chain1 (str) – The chain used for the residue specified by rank_row when measuring distances
- chain2 (str) – The chain used for the residue specified by rank_col when measuring distances
-
buildSeqidMatrix(msa, turbo=True)[source]¶ Returns sequence identity matrix for msa.
By default, turbo mode, which uses memory as large as the MSA array itself but runs four to five times faster, will be used. If memory allocation fails, the implementation will fall back to slower and memory efficient mode.
-
uniqueSequences(msa, seqid=0.98, turbo=True)[source]¶ Returns a boolean array marking unique sequences in msa. A sequence sharing sequence identity of seqid or more with another sequence coming before itself in msa will have a True value in the array.
By default, turbo mode, which uses memory as large as the MSA array itself but runs four to five times faster, will be used. If memory allocation fails, the implementation will fall back to slower and memory efficient mode.
-
buildOMESMatrix(msa, ambiguity=True, turbo=True, **kwargs)[source]¶ Returns OMES (Observed Minus Expected Squared) covariance matrix calculated for msa, which may be an
MSAinstance or a 2D NumPy character array. OMES is defined as:(N_OBS - N_EX)^2 (f_i,j - f_i * f_j)^2 OMES_(i,j) = sum(------------------) = N * sum(-----------------------) N_EX f_i * f_jImplementation is case insensitive and handles ambiguous amino acids as follows:
- B (Asx) count is allocated to D (Asp) and N (Asn)
- Z (Glx) count is allocated to E (Glu) and Q (Gln)
- J (Xle) count is allocated to I (Ile) and L (Leu)
- X (Xaa) count is allocated to the twenty standard amino acids
- Joint probability of observing a pair of ambiguous amino acids is allocated to all potential combinations, e.g. probability of XX is allocated to 400 combinations of standard amino acids, similarly probability of XB is allocated to 40 combinations of D and N with the standard amino acids.
Selenocysteine (U, Sec) and pyrrolysine (O, Pyl) are considered as distinct amino acids. When ambiguity is set False, all alphabet characters as considered as distinct types. All non-alphabet characters are considered as gaps.
By default, turbo mode, which uses memory as large as the MSA array itself but runs four to five times faster, will be used. If memory allocation fails, the implementation will fall back to slower and memory efficient mode.
-
buildSCAMatrix(msa, turbo=True, **kwargs)[source]¶ Returns SCA matrix calculated for msa, which may be an
MSAinstance or a 2D Numpy character array.Implementation is case insensitive and handles ambiguous amino acids as follows:
- B (Asx) count is allocated to D (Asp) and N (Asn)
- Z (Glx) count is allocated to E (Glu) and Q (Gln)
- J (Xle) count is allocated to I (Ile) and L (Leu)
- X (Xaa) count is allocated to the twenty standard amino acids
- Joint probability of observing a pair of ambiguous amino acids is allocated to all potential combinations, e.g. probability of XX is allocated to 400 combinations of standard amino acids, similarly probability of XB is allocated to 40 combinations of D and N with the standard amino acids.
Selenocysteine (U, Sec) and pyrrolysine (O, Pyl) are considered as distinct amino acids. When ambiguity is set False, all alphabet characters as considered as distinct types. All non-alphabet characters are considered as gaps.
By default, turbo mode, which uses memory as large as the MSA array itself but runs four to five times faster, will be used. If memory allocation fails, the implementation will fall back to slower and memory efficient mode.
-
buildDirectInfoMatrix(msa, seqid=0.8, pseudo_weight=0.5, refine=False, **kwargs)[source]¶ Returns direct information matrix calculated for msa, which may be an
MSAinstance or a 2D Numpy character array.Sequences sharing sequence identity of seqid or more with another sequence are regarded as similar sequences for calculating their weights using
calcMeff().pseudo_weight are the weight for pseudo count probability.
Sequences are not refined by default. When refine is set True, the MSA will be refined by the first sequence and the shape of direct information matrix will be smaller.
-
calcMeff(msa, seqid=0.8, refine=False, weight=False, **kwargs)[source]¶ Returns the Meff for msa, which may be an
MSAinstance or a 2D Numpy character array.Since similar sequences in an msa decreases the diversity of msa, Meff gives a weight for sequences in the msa.
For example: One sequence in MSA has 5 other similar sequences in this MSA(itself included). The weight of this sequence is defined as 1/5=0.2. Meff is the sum of all sequence weights. In another word, Meff can be understood as the effective number of independent sequences.
Sequences sharing sequence identity of seqid or more with another sequence are regarded as similar sequences to calculate Meff.
Sequences are not refined by default. When refine is set True, the MSA will be refined by the first sequence.
The weight for each sequence are returned when weight is True.
-
buildPCMatrix(msa, turbo=False, **kwargs)[source]¶ Returns PC matrix calculated for msa, which may be an
MSAinstance or a 2D Numpy character array.Implementation is case insensitive and handles ambiguous amino acids as follows:
- B (Asx) count is allocated to D (Asp) and N (Asn)
- Z (Glx) count is allocated to E (Glu) and Q (Gln)
- J (Xle) count is allocated to I (Ile) and L (Leu)
- X (Xaa) count is allocated to the twenty standard amino acids
- Joint probability of observing a pair of ambiguous amino acids is allocated to all potential combinations, e.g. probability of XX is allocated to 400 combinations of standard amino acids, similarly probability of XB is allocated to 40 combinations of D and N with the standard amino acids.
Selenocysteine (U, Sec) and pyrrolysine (O, Pyl) are considered as distinct amino acids. When ambiguity is set False, all alphabet characters as considered as distinct types. All non-alphabet characters are considered as gaps.
-
buildMSA(sequences, title='Unknown', labels=None, **kwargs)[source]¶ Aligns sequences with clustalw or clustalw2 and returns the resulting MSA.
Parameters: - sequences (
Atomic,MSA,ndarray, str) – a file, MSA object or a list or array containing sequences as Atomic objects withgetSequence()or Sequence objects or strings. If strings are used then labels must be provided usinglabels - title (str) – the title for the MSA and it will be used as the prefix for output files.
- labels (list) – a list of labels to go with the sequences
- align (str) – whether to align the sequences default True
- method – alignment method, one of either ‘global’ (biopython.pairwise2.align.globalms), ‘local’ (biopython.pairwise2.align.localms), clustalw(2), or another software in your path. Default is ‘clustalw’
- sequences (
-
showAlignment(alignment, **kwargs)[source]¶ Prints out an alignment as sets of short rows with labels.
Parameters: - alignment – any object with aligned sequences
- row_size (int) – the size of each row default 60
- indices (
ndarray, list) – a set of indices for some or all sequences that will be shown above the relevant sequences - index_start (int) – how far along the alignment to start putting indices default 0
- index_stop (int) – how far along the alignment to stop putting indices default the point when the shortest sequence stops
- labels (list) – a list of labels
-
alignTwoSequencesWithBiopython(seq1, seq2, **kwargs)[source]¶ Easily align two sequences with Biopython’s globalms or localms. Returns an MSA and indices for use with
showAlignment().Alignment parameters can be provided as keyword arguments. Default values are as originally set in the proteins.compare module, but now found in utilities.seqtools.
Parameters: - match (int) – a positive integer, used to reward finding a match
- mismatch (int) – a negative integer, used to penalise finding a mismatch
- gap_opening (int) – a negative integer, used to penalise opening a gap
- gap_extension (int) – a negative integer, used to penalise extending a gap
- method (str) – method for pairwise2 alignment. Possible values are ‘local’ and ‘global’
-
alignSequenceToMSA(seq, msa, **kwargs)[source]¶ Align a sequence from a PDB or Sequence to a sequence from an MSA and create two sets of indices.
The sequence from the MSA (seq), the alignment and the two sets of indices are returned.
The first set (indices) maps the residue numbers in the PDB to the reference sequence. The second set (msa_indices) indexes the reference sequence in the msa and is used for retrieving values from the first indices.
Parameters: - seq (
Atomic,Sequence, str) – an object with an associated sequence string or a sequence string itself - msa (
MSA) – a multiple sequence alignment - label (str) – a label for a sequence in msa or a PDB ID
msa.getIndex(label)must return a sequence index - chain (str) – which chain from pdb to use for alignment, default is None,
which does no selection on seq. This value will be ignored if seq is
not an
Atomicobject.
Parameters for Biopython
pairwise2alignments can be provided as keyword arguments. Default values are originally fromproteins.comparemodule, but now found inutilities.seqtools.Parameters: - match (int) – a positive integer, used to reward finding a match
- mismatch (int) – a negative integer, used to penalise finding a mismatch
- gap_opening (int) – a negative integer, used to penalise opening a gap
- gap_extension (int) – a negative integer, used to penalise extending a gap
- method (str) – method for pairwise2 alignment.
Possible values are
"local"and"global"
- seq (
-
alignSequencesByChain(PDBs, **kwargs)[source]¶ Runs
buildMSA()for each chain and optionally joins the results. Returns either a singleMSAor a dictionary containing anMSAfor each chain.Parameters:
-
trimAtomsUsingMSA(atoms, msa, **kwargs)[source]¶ This function uses
alignSequenceToMSA()and has the same kwargs.Parameters: