Probe Grid Calculation¶
When simulations are complete, you need to perform grid calculations using the following interface:
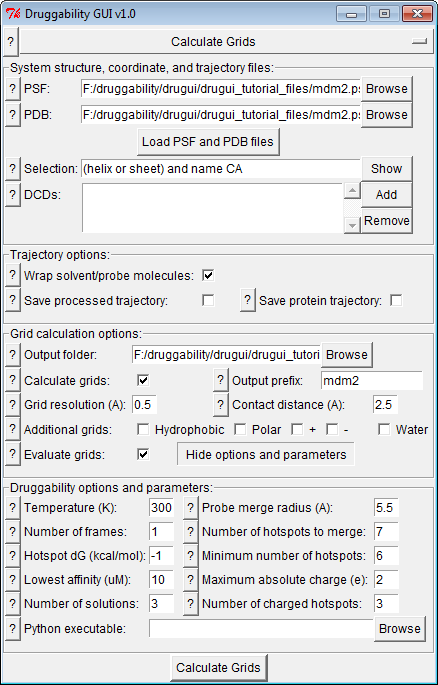
Input Files¶
Input files are prefix.psf and prefix.pdb files,
and simulation trajectory files, e.g. prefix_sim/sim.dcd.
If you performed multiple simulations for the same system, you can
include all productive simulation trajectories in grid calculations.
Ideally, trajectories from equilibration simulations should not be
included in grid calculations. Selection specifies atoms
used to align the target conformations in trajectory frames.
Note
Selection is an important input for grid calculations. If the binding site move internally when all atoms of the protein are used for alignment, you may want to restrict the alignment to binding site residues excluding those that are mobile. This will help capturing probe enrichment at a binding site properly. If there are multiple binding sites that move internally in a protein, it would be better to analyze those sites one by one.
Options & Parameters¶
For probe enrichment (or occupancy) grid calculations, probe and solvent molecules needs to be wrapped. If you like and have enough disk space, you may write trajectory frames after they are wrapped. This may be good for later visualization and movie making purposes.
By default, grids will be calculated for different probe types using their central carbon atoms. These grids will be merged in druggability analysis. Default grid resolution 0.5 Å has been found to capture probe locations ideally.
For visualization purposes, you may select to output occupancy grids for hydrophobic, polar, charged, and water atoms.
Grids can be visualized using Chimera Volume Viewer or VMD_.
You may also select to evaluate grids immediately after their calculation. Details of this step is discussed in the next part.
Output Files¶
Output files are occupancy grids for each probe type, e.g.
prefix_IPRO.dx, and selected atom types.