Multimeric Structures¶
In this part, first we will build an ensemble of
HIV Reverse Transcriptase (RT), which is a heterodimer. The ensemble
can be conveniently built by buildPDBEnsemble() automatically, but we
will build it step by step first for a better understanding of the process.
Then we will show how to use buildPDBEnsemble() to build ensemble in
one line. Then we will perform PCA on the ensemble.
Input and Parameters¶
First, we make necessary imports:
In [1]: from prody import *
In [2]: from pylab import *
In [3]: ion()
Reference structure¶
We set the name of the protein/dataset (a name without a white space is preferred) and also reference structure id and chain identifiers:
In [4]: name = 'HIV-RT' # dataset name
In [5]: ref_pdb = '1dlo' # reference PDB file
In [6]: ref_chids = 'AB' # reference chain identifiers
Parameters¶
The following parameters are for comparing two structures to determine matching chains.
In [7]: sequence_identity = 94
In [8]: sequence_coverage = 85
Chains from two different structures will be paired if they share 94% sequence identity and the aligned part of the sequences cover 85% of the longer sequence.
Structures¶
We are going to use the following list of structures:
In [9]: pdb_ids = ['3kk3', '3kk2', '3kk1', '1suq', '1dtt', '3i0r', '3i0s', '3m8p',
...: '3m8q', '1jlq', '3nbp', '1klm', '2ops', '2opr', '1s9g', '2jle',
...: '1s9e', '1jla', '1jlc', '1jlb', '1jle', '1jlg', '1jlf', '3drs',
...: '3e01', '3drp', '1hpz', '3ith', '1s1v', '1s1u', '1s1t', '1ep4',
...: '3klf', '2wom', '2won', '1s1x', '2zd1', '3kle', '1hqe', '1n5y',
...: '1fko', '1hnv', '1hni', '1hqu', '1iky', '1ikx', '1t03', '1ikw',
...: '1ikv', '1t05', '3qip', '3jsm', '1c0t', '1c0u', '2ze2', '1hys',
...: '1rev', '3dle', '1uwb', '3dlg', '3qo9', '1tv6', '2i5j', '3meg',
...: '3mee', '3med', '3mec', '3dya', '2be2', '2opp', '3di6', '1tl3',
...: '1jkh', '1sv5', '1tl1', '1n6q', '2rki', '1tvr', '3klh', '3kli',
...: '1dtq', '1bqn', '3klg', '1bqm', '3ig1', '2b5j', '1r0a', '3dol',
...: '1fk9', '2ykm', '1rtd', '1hmv', '3dok', '1rti', '1rth', '1rtj',
...: '1dlo', '1fkp', '3bgr', '1c1c', '1c1b', '3lan', '3lal', '3lam',
...: '3lak', '3drr', '2rf2', '1rt1', '1j5o', '1rt3', '1rt2', '1rt5',
...: '1rt4', '1rt7', '1rt6', '3lp1', '3lp0', '2iaj', '3lp2', '1qe1',
...: '3dlk', '1s1w', '3isn', '3kjv', '3jyt', '2ban', '3dmj', '2vg5',
...: '1vru', '1vrt', '1lw2', '1lw0', '2ic3', '3c6t', '3c6u', '3is9',
...: '2ykn', '1hvu', '3irx', '2b6a', '3hvt', '1tkz', '1eet', '1tkx',
...: '2vg7', '2hmi', '1lwf', '1tkt', '2vg6', '1s6p', '1s6q', '3dm2',
...: '1lwc', '3ffi', '1lwe']
...:
A predefined set of structures will be used, but an up-to-date list can be obtained by blast searching PDB. See Homologous Proteins and Blast Search PDB examples.
Set reference¶
Now we set the reference chains that will be used for compared to the structures in the ensemble and will form the basis of the structural alignment.
# Parse reference structure
In [10]: reference_structure = parsePDB(ref_pdb, subset='calpha')
# Get the reference chain from this structure
In [11]: reference_hierview = reference_structure.getHierView()
In [12]: reference_chains = [reference_hierview[chid] for chid in ref_chids]
In [13]: reference_chains
Out[13]:
[<Chain: A from 1dlo_ca (556 residues, 556 atoms)>,
<Chain: B from 1dlo_ca (415 residues, 415 atoms)>]
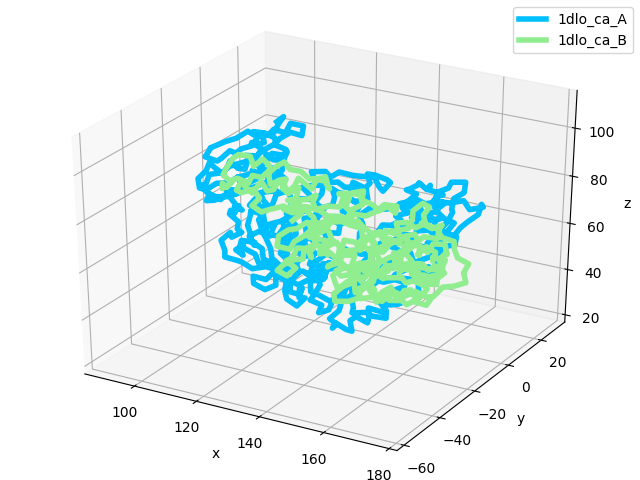
Chain A is the p66 subunit, and chain B is the p51 subunit of HIV-RT. Let’s take a quick look at that:
In [14]: showProtein(reference_structure);
In [15]: legend();
Prepare Ensemble¶
We handle an ensemble of heterogeneous conformations using
PDBEnsemble objects, so let’s instantiate one:
In [16]: ensemble = PDBEnsemble(name)
We now combine the reference chains and set the reference coordinates of the ensemble.
In [17]: reference_chain = reference_chains[0] + reference_chains[1]
In [18]: ensemble.setAtoms(reference_chain)
In [19]: ensemble.setCoords(reference_chain.getCoords())
Coordinates of the reference structure are set as the coordinates of the ensemble onto which other conformations will be superposed.
We can also start a log file using startLogfile().
Screen output will be saved in this file, and can be
used to check if structures are added to the ensemble as expected.
In [20]: startLogfile(name)
Let’s also start a list to keep track of PDB files that are not added to the ensemble:
In [21]: unmapped = []
Now, we parse the PDB files one by one and add them to the ensemble:
In [22]: for pdb in pdb_ids:
....: structure = parsePDB(pdb, subset='calpha', model=1)
....: atommaps = []
....: for reference_chain in reference_chains:
....: mappings = mapOntoChain(structure, reference_chain,
....: seqid=sequence_identity,
....: coverage=sequence_coverage)
....: if len(mappings) == 0:
....: print('Failed to map', pdb)
....: break
....: atommaps.append(mappings[0][0])
....: if len(atommaps) != len(reference_chains):
....: unmapped.append(pdb)
....: continue
....: atommap = atommaps[0] + atommaps[1]
....: ensemble.addCoordset(atommap, weights=atommap.getFlags('mapped'))
....:
In [23]: ensemble
Out[23]: <PDBEnsemble: HIV-RT (155 conformations; 971 atoms)>
In [24]: ensemble.iterpose()
In [25]: saveEnsemble(ensemble)
Out[25]: 'HIV-RT.ens.npz'
We can now close the logfile using closeLogfile():
In [26]: closeLogfile(name)
Let’s check which structures, if any, are not mapped (added to the ensemble):
In [27]: unmapped
Out[27]: []
We can write the aligned conformations into a PDB file as follows:
In [28]: writePDB(name + '.pdb', ensemble)
Out[28]: 'HIV-RT.pdb'
This file can be used to visualize the aligned conformations in molecular graphics software.
This is a heterogeneous dataset, i.e. many structures have missing residues. We want to make sure that we include residues in PCA analysis if they are resolved more than 94% of the time.
We can check this using the calcOccupancies() function:
In [29]: calcOccupancies(ensemble, normed=True).min()
Out[29]: 0.3096774193548387
This shows that some residues were resolved in only 24% of the dataset. We trim the ensemble to contain residues resolved in more than 94% of the ensemble:
In [30]: ensemble = trimPDBEnsemble(ensemble, occupancy=0.94)
After trimming, another round of iterative superposition may be useful:
In [31]: ensemble.iterpose()
In [32]: ensemble
Out[32]: <PDBEnsemble: HIV-RT (155 conformations; selected 908 of 971 atoms)>
In [33]: saveEnsemble(ensemble)
Out[33]: 'HIV-RT.ens.npz'
Use buildPDBEnsemble Function¶
As mentioned at the beginning, the ensemble can be also built by
buildPDBEnsemble() in several lines of code:
In [34]: unmapped = []
In [35]: prot = parsePDB('1dlo', subset='ca', model=1)
In [36]: pdbs = parsePDB(pdb_ids, subset='ca', model=1)
In [37]: ensemble = buildPDBEnsemble(pdbs, ref=prot, title='HIV-RT', labels=pdb_ids,
....: seqid=94, coverage=85, occupancy=0.94, unmapped=unmapped)
....:
In [38]: ensemble
Out[38]: <PDBEnsemble: HIV-RT (162 conformations; selected 893 of 971 atoms)>
Perform PCA¶
Once the ensemble is ready, performing PCA is 3 easy steps:
In [39]: pca = PCA(name)
In [40]: pca.buildCovariance(ensemble)
In [41]: pca.calcModes()
The calculated data can be saved as a compressed file using saveModel()
In [42]: saveModel(pca)
Out[42]: 'HIV-RT.pca.npz'
Plot results¶
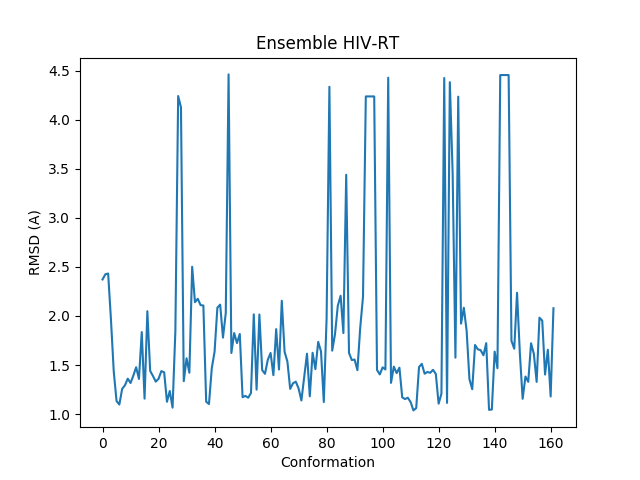
Let’s plot RMSD from the average structure:
In [43]: plot(calcRMSD(ensemble));
In [44]: xlabel('Conformation');
In [45]: ylabel('RMSD (A)');
In [46]: title(ensemble);
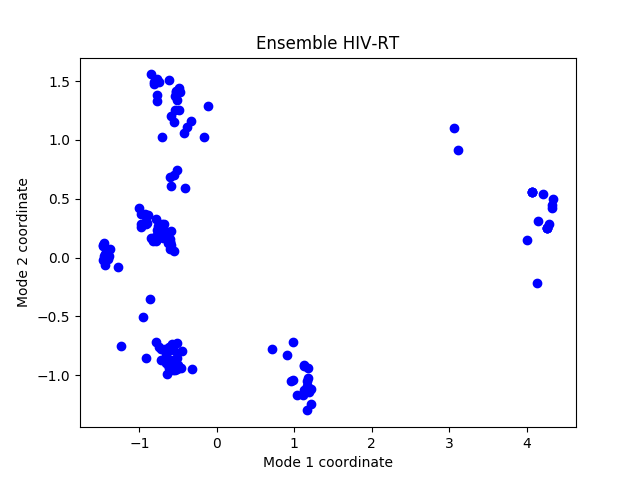
Let’s show a projection of the ensemble onto PC1 and PC2:
In [47]: showProjection(ensemble, pca[:2]);
In [48]: title(ensemble);
Only some of the ProDy plotting functions are shown here. A complete list can be found in Dynamics Analysis module.