Pharmmaker Tutorial
Bahar lab, University of Pittsburgh
Download:
Pharmmaker program: http://prody.csb.pitt.edu/tutorials/pharmmaker/pharmmaker.zip
DruGUI results: http://prody.csb.pitt.edu/tutorials/pharmmaker/drugui_results.zip
PDF version of this tutorial: http://prody.csb.pitt.edu/tutorials/pharmmaker/pharmmaker_tutorial.pdf
Introduction
Based on druggability simulations, we define a druggable pocket, collect snapshots from the trajectory with probe molecules in this pocket, and build pharmacophore models as shown below.
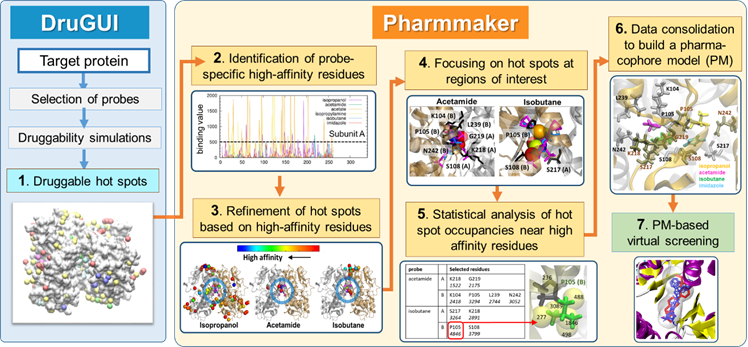
Figure 1. Pharmmaker workflow to construct pharmacophore models (PMs) based on druggability simulations. Pharmmaker works in conjunction with DruGUI developed for generating druggability trajectories and identifying druggable hot spots (blue box). The output from DruGUI is subjected to Steps 2-6 (yellow box), to release a PM that is used (in step 7) for virtual screening of libraries of compounds.
In the tutorial directory, there are three directories: drugui-simulation, drugui-analysis, and pharmmaker. The drugui-simulation directory includes outputs form druggability simulation, the drugui-analysis directory includes outputs from druggability analysis, and the pharmmaker directory includes programs to build pharmacophore model using the outputs in the two directories. In this tutorial, we explain how to use Pharmmaker. You will need VMD and a shell (Linux/Mac).
We also provide an optional python file to aid with the analysis. To use this file, you will need to install our ProDy API as well as NumPy and SciPy. Instructions for how to download and install ProDy can be found at http://prody.csb.pitt.edu/downloads/ and NumPy and SciPy can be installed in similar ways. We recommend using the Anaconda Python distribution, which handles libraries well and already includes many of them as well as some useful interactive features.
1. Druggability simulation [directories drugui-simulation and drugui- analysis]
In the directory drugui-simulation, there are two files protein-probe.pdb and protein-probe.dcd. The file protein-probe.pdb is the initial structure for simulation, and protein-probe.dcd is the output of the simulation including snapshots for 40 ns. Here, we excluded water molecules and ions to reduce the size of the tutorial directory. These two files were also used as inputs for druggability analysis, which would be the next step (already performed here). We don’t explain here how to make input files for simulations and run such simulations. Details can be found at http://prody.csb.pitt.edu/tutorials/drugui_tutorial/
2. Druggability analysis [directory drugui- analysis]
In the directory drugui-analysis, there are two files dg_protein_heavyatoms.pdb and dg_all_hotspots.pdb. The file dg_protein_heavyatoms.pdb is the same as the protein-probe.pdb but only including protein heavy atoms, which are used for this analysis. The file dg_all_hotspots.pdb is the output of the analysis. Again, we don’t explain how to analyze druggability with DruGUI. Details can again be found online at http://prody.csb.pitt.edu/tutorials/drugui_tutorial/
If you check these files using VMD, you will see the following:
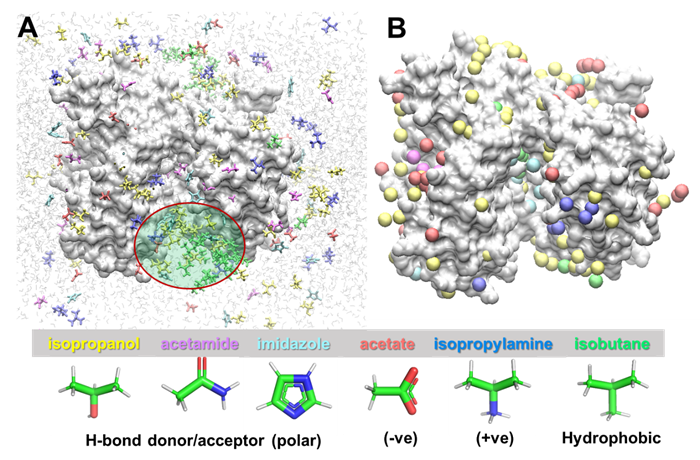
Figure 2. Druggability simulations. A. A snapshot of the simulated system. An LBD dimer is shown in silver surface representation and probe molecules are shown as sticks colored by types. Six types of probes (isopropanol (yellow), acetamide (magenta), imidazole (cyan), acetate (red), isopropylamine (blue) and isobutane (green)) were used, and their structures and features are indicated at the bottom. Water molecules are shown as shaded light gray lines in the background. The entrance to the dimer interface (enclosed in a red ellipse) is crowded by probes. B. Hot spots from the druggability analysis. Hot spots are voxels in 3D space which are highly occupied by probe molecules, showing which region of protein is highly druggable. Hot spots are obtained for each probe molecule type and are displayed as balls in the same color as the probe.
3. Pharmmaker: Building pharmacophore models
In the directory pharmmaker, there are three files run_interface_highaffresid.py, run_hotspotsNearHighAffResids.sh and run_snapshot.sh and a directory CORE. These three files include input parameters and command lines to run the core Pharmmaker programs found in the CORE directory.
Before we start, we suggest you create a new directory for outputs and go to this directory. We explain how to use Pharmmaker assuming you are in this directory.
3.1. High affinity residues
In the pharmmaker/CORE directory, there is one file for finding high affinity residues called highaffresid.tcl, which is run using VMD. We can do this directly or we can use a shell or python script to give it more sophisticated inputs. We’ll first describe the direct way and then show you how to use the python script we provided, which allows us to define the dimer interface as a region of interest.
To run the command with VMD, we call VMD as usual but with a few significant differences. At the beginning of the command, you’ll notice env VMDARGS=‘text with blanks’ to tell VMD that we want to provide it with arguments, which are text elements separated with blanks, and we mark where they start with the -args flag. We use the -dispdev flag to tell it we want to run VMD in text mode and the -e flag to tell it we want to execute our script whose name we then provide. The arguments we provide are the path to a PDB and DCD file coming from a simulation prepared with DruGUI, a chain identifier, a probe type, and first and last residue numbers.
[outputs] env VMDARGS='text with blanks' vmd -dispdev text -e ../pharmmaker/CORE/highaffresid.tcl -args ../drugui-simulation/protein-probe.pdb ../drugui-simulation/protein-probe.dcd A IPRO 105 110
In this case, we ask it to analyze interactions of chain A with probe IPRO (isopropylamine) and only include residues 105 to 110 inclusive in the analysis to make it quick and easy. We also use PDB and DCD files that only contain protein and probe atoms without water and ions.
We can also provide it with comma-separated lists of multiple chain IDs and probe types as follows.
[outputs] env VMDARGS='text with blanks' vmd -dispdev text -e ../pharmmaker/CORE/highaffresid.tcl -args ../drugui-simulation/protein-probe.pdb ../drugui-simulation/protein-probe.dcd A,B IPRO,ACTT 105 110
We can see more information about the options if we provide it with the single argument help:
[outputs] env VMDARGS='text with blanks' vmd -dispdev text -e highaffresid.tcl -args help
We can also use it in a more complicated fashion from a Python script that uses our ProDy API to provide it with a longer list of residues to define a region of interest more easily. The example script (also found in the pharmmaker directory) is uses the dimer interface as described in the manuscript.
[outputs] python ../pharmmaker/run_interface_highaffresid.py ../drugui-simulation/protein-probe.pdb ../drugui-simulation/protein-probe.dcd
Output files such as out-A-IPRO.dat are generated, which contain binding values as shown below:
=========== out-A-IPRO.dat ===========
5 1196.5305490795165
6 270.57834273413806
7 94.91752249348069
8 0
~~
=========== out-A-IPRO.dat ===========
In the file above, the first column is residue number and the second column the binding value with IPRO. Repeating this for all probes and chains, we can plot binding value graphs:
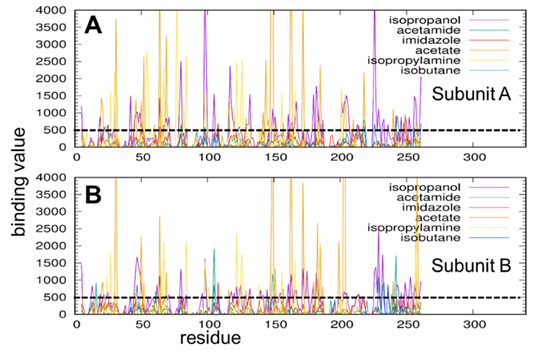
Figure 3. High affinity residues of AMPAR GluA2 LBD dimer interacting with different types of probe molecules. A-B. Binding score profile for LBD subunits A and B (labeled), evaluated for each probe molecule (curves in different colors). Binding score is as described in the text. Residues with score above 500/Å2 (shown in dashed line) are identified as high affinity residues for each type of probe. The residue numbers refer to those in the examined PDB file, corresponding to the isolated LBD (not the full receptor).
From the graph, we select high affinity residues with a binding value larger than a criterion. Here, we chose the criterion as 500.
The program also uses a value of 500 by default (which is good for this size trajectory) and returns files such as out-A-IPRO-highaffresids.dat, which contain list of high affinity residues:
=========== out-A-IPRO-highaffresids.dat ===========
218 243 244 ~~
=========== out-A-IPRO-highaffresids.dat ===========
We also obtain a summary file called highaffresid.dat, which contains information about the high affinity residues with binding values above the cutoff sorted by probe type and chain as follows:
=========== highaffresid.dat ===========
ACTT A 218 ~~
~~~
IMID A 98 108 ~~
IPRO A 218 243 244 ~~
~~~
=========== highaffresid.dat ===========
3.2. Hot spots near high affinity residues
In the pharmmaker/CORE directory, there is a file called hotspotsNearHighAffResids.sh, which finds hot spots near high affinity residues as the name says. It can be run as follows:
[outputs] bash ../pharmmaker/CORE/hotspotsNearHighAffResids.sh A IBTN 8 ../drugui-analysis/dg_all_hotspots.pdb ../drugui-analyis/dg_protein_heavyatoms.pdb .
The arguments after the filename are chain ID, probe type, cutoff distance for hotspots being near residues in Å, paths for PDB files containing hotspots and protein heavy atoms from DruGUI analysis, and the directory containing the lists of high affinity residues (the dot means the current working directory and finds the files from above based on the chain ID and probe type).
This script can be run multiple times for multiple chains and probe types. The file run_hotspotsNearHighAffResids.sh in the main pharmmaker directory does this. It takes as input a list of probes and a list of chains that is generated by the previous step and then the remaining inputs are the same.
[outputs] bash ../pharmmaker/run_hotspotsNearHighResids.sh probe-list.dat chain-list.dat 8 ../drugui-analysis/dg_all_hotspots.pdb ../drugui-analyis/dg_protein_heavyatoms.pdb .
The output file highAffHotspots.pdb looks something like this:
=========== highAffHotspots.pdb ===========
ATOM 14 C2 ACTTA 5 11.256 5.981 -2.757 1.00 -2.05 M
ATOM 54 C2 ACTTA 5 10.756 3.481 -1.757 1.00 -1.52 M
ATOM 169 C2 ACTTA 5 10.756 7.481 -4.757 1.00 -1.11 M
ATOM 176 C2 ACTTA 5 10.256 9.981 -4.757 0.99 -1.09 M
ATOM 8 C2 ACTTA 5 11.756 9.481 -6.757 1.00 -2.13 M
~~~
ATOM 43 C2 IBTNA 3 -1.244 2.981 -1.757 0.67 -1.66 M
ATOM 152 C2 IBTNA 3 -2.244 5.481 -0.757 0.72 -1.13 M
ATOM 4 C2 IBTNA 3 -0.244 0.481 -1.257 0.86 -2.31 M
ATOM 39 C2 IBTNA 3 0.756 0.481 -3.757 0.72 -1.67 M
ATOM 55 C2 IBTNA 3 -0.744 3.981 -5.757 0.54 -1.48 M
ATOM 117 C2 IBTNA 3 -1.244 -2.019 -0.757 0.72 -1.23 M
~~~
ATOM 13 C IMIDA 3 -1.744 -4.519 -0.257 0.49 -2.07 M
ATOM 91 C IMIDA 3 1.756 -9.019 4.243 0.98 -1.31 M
ATOM 143 C IMIDA 47 3.256 -0.019 6.243 0.59 -1.17 M
~~~
=========== highAffHotspots.pdb ===========
It is a selection of PDB lines for hot spots of each probe type that are near high affinity residues. The B-factor column has the affinity of each hotspot and each probes type has hot spots that are ordered by this affinity value.
3.3. Focusing on hot spots at regions of interest
At this point, we recommend you to inspect the output residues and hotspots again using VMD. You may see that some high affinity residues and hotspots are not really in a region of interest.
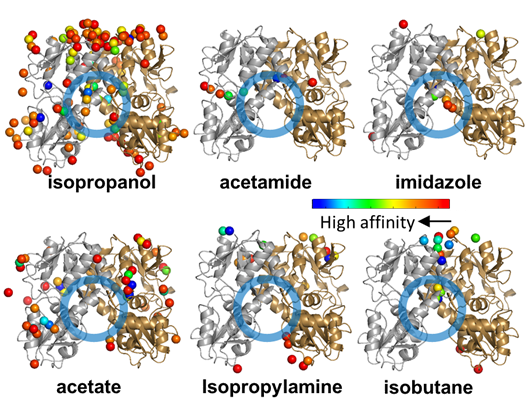
Figure 4. Hot spots near high affinity residues. Spatial distribution of probe molecules near high affinity residues, shown separately for each type of probe, color-coded by affinity. A hot spot for isopropanol binding, also highlighted as a region of interest in Fig 2A, is indicated by the blue circle. Note that this region also binds imidazole, acetamide and isobutane, but not acetate and isopropylamine (see Fig 5).
At a first glance, we immediately see that there are no hot spots for acetamide (ACAM) and isopropylamine (IPAM) in our region of interest, and many of the isopropanol (IPRO) and isobutane (IBTN) hot spots are at the top of the dimer in the vicinity of the interface and not in it. Next, we therefore remove the ACAM and IPAM hot spots and high affinity residues from the respective files (after copying them if you want to keep the results of the original analysis).
We can also perform a more detailed analysis and remove other hot spots and high affinity residues based on their atom or residue numbers. A good example of this for residues is Gln244, which projects away from the main dimer interface between the upper (D1) lobes and sometimes reaches down towards the lower (D2) lobe.
Such an analysis reveals that a much number of residues and hot spots are truly in a meaningful region as interest (see below):
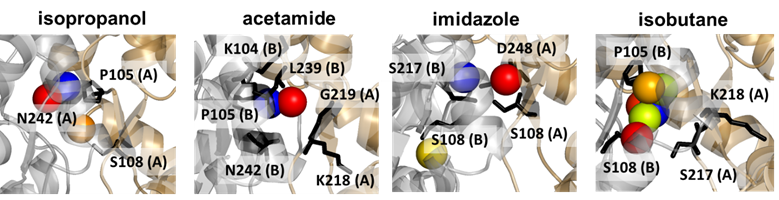
Figure 5. Probes and high affinity residues in our region of interest. This region binds four types of probes. The corresponding poses and coordinating residues are displayed for each type of probe (color-coded by binding score) in the four diagrams.
We therefore suggest modifying the highaffresid.dat and highAffHotspots.pdb to look something like the following before preceding to the next step:
=========== highaffresid.dat ===========
ACAM B 104 105 239 242
IBTN A 217 218
IBTN B 105 108
IMID A 108 248
IMID B 108 217
IPRO A 105 108 242
=========== highaffresid.dat ===========
In this file, write all selected high affinity residues in our region of interest.
=========== highAffHotspots.pdb ===========
ATOM 1 C IBTNA 1 -0.244 0.481 -1.257 0.86 -2.31 M
ATOM 2 C IBTNA 2 0.756 0.481 -3.757 0.72 -1.67 M
ATOM 3 C IBTNA 3 -1.244 2.981 -1.757 0.67 -1.66 M
ATOM 4 C IBTNA 4 -0.744 3.981 -5.757 0.54 -1.48 M
ATOM 5 C IBTNA 5 -1.244 -2.019 -0.757 0.72 -1.23 M
ATOM 6 C IBTNA 6 -2.244 5.481 -0.757 0.72 -1.13 M
ATOM 1 C IPROA 1 -1.744 -8.519 -3.757 1.00 -1.97 M
ATOM 2 C IPROA 2 -0.244 -6.519 0.743 0.50 -1.70 M
ATOM 3 C IPROA 3 -3.744 -6.519 -3.257 0.97 -1.61 M
ATOM 1 C ACAMA 1 0.756 1.981 -5.757 0.98 -2.30 M
ATOM 2 C ACAMA 2 3.256 1.481 -5.757 0.99 -1.03 M
ATOM 13 C IMIDA 3 -1.744 -4.519 -0.257 0.49 -2.07 M
ATOM 91 C IMIDA 3 1.756 -9.019 4.243 0.98 -1.31 M
ATOM 2 C IMIDA 2 -4.244 5.981 1.243 0.80 -1.58 M
=========== highAffHotspots.pdb ===========
In this file, write all selected hot spots in our region of interest.
3.4 Snapshots with probes at the hot spots and high affinity residues
Again, there are core program files to do this (snapshot2.tcl and snapshot2.tcl) and a run script run_snapshot.sh for handling their inputs and outputs.
[outputs] bash ../pharmmaker/run_snapshot.sh ../drugui-simulation/protein-probe.pdb ../drugui-simulation/protein-probe.dcd
Output files are generated in the snapshot directory and subdirectories inside it. For example, for residue 105 of subunit A with probe isopropanol, there is a directory z.A.105.IPRO with the following files in it: outfr-1.dat, outfr-2.dat, outfr-3.dat, outfr-count, out-detail-res-1.dat, out-detail-res-2.dat, out-detail-res-3.dat, out-detail-hs-1.dat, out-detail-hs-2.dat, out-detail-hs-3.dat, and outfr.dat.
=========== outfr-1.dat ===========
1191
1192
1196
~~
=========== outfr-1.dat ===========
In the file above, the first column is frame number where there is a probe of isopropanol interacting with residue 105 and also is located in the hotspot number 1.
In this case, we wrote three hotspots for isopropanol in hotspot.pdb, and its number 1 is the one with the highest affinity in 9th column in hotspot.pdb. The other files outfr-2.dat and outfr-3.dat are the equivalent results for the two other hotspots, here hot spot numbers are defined by in affinity order starting with the highest affinity.
=========== outfr-count ===========
725 z.A.105.IPRO/out-1.dat
462 z.A.105.IPRO/out-2.dat
660 z.A.105.IPRO/out-3.dat
=========== outfr-count ===========
In this file, the first column is total frame numbers for each case and the second column is a path to the corresponding file above. The first line means that there are 725 snapshots where there is a probe interacting with residue 105 and being located at hot spot number 1. The following lines then refer to the subsequent hotspots with the number of snapshots going down as expected from the lower affinity.
The next two files provide details about probes near high affinity residues and which of those probes are near hotspots. These two pieces of information are essential for deciding which snapshots to use to generate a pharmacophore model in the final step.
out-detail-res-1.dat is below:
=========== out-detail-res-1.dat ===========
1191 1590 PRO 105 C 9428 IPRO 7 OH2 3.7905426025390625
1191 1591 PRO 105 O 9418 IPRO 7 C2 3.4408786296844482
1191 1591 PRO 105 O 9424 IPRO 7 C3 3.641240119934082
1191 1591 PRO 105 O 9428 IPRO 7 OH2 2.6508584022521973
1192 1578 PRO 105 N 9428 IPRO 7 OH2 3.7221927642822266
~~
=========== out-detail-res-1.dat ===========
In the file above, we have the following fields: snapshot number, 2 x (atom serial number, residue name, residue number, atom name) corresponding to protein and probe atoms, and their distances.
The other files out-detail-res-2.dat, out-detail-res-3.dat are equivalent results with the two other hotspots.
out-detail-hs-1.dat is below:
=========== out-detail-hs-1.dat ===========
1191 IPRO 7 C3 IPRO 1 1.3570990562438965
1192 IPRO 7 C3 IPRO 1 1.3819466829299927
1196 IPRO 7 C3 IPRO 1 1.4686346054077148
~~
=========== out-detail-hs-1.dat ===========
In the file above, we have the following fields: snapshot number, (probe name, probe number, atom name) for probe, and (probe name, number) for hotspot, and their distance. The other out-detail-hs-2.dat, out-detail-hs-3.dat are equivalent results with the two other hotspots.
Finally, outfr.dat is below:
=========== outfr.dat ===========
frame 601
frame 603
frame 606
frame 607
=========== outfr.dat ===========
In the file above, the second column is frame numbers where there is a probe interacting with residue 105 and also being located at any of the three hotspots. Therefore, the total number of lines in this file is 1769, which is less than 1847, the sum of the frame numbers in out-1.dat, out-2.dat, and out-3.dat (725 + 462 + 660), because of shared snapshots among the three hotspots.
We explained the case for residue 105 of subunit A and probe isopropanol (see Fig 5), but there are directories like z.A.105.IPRO for all the cases written in the input file highaffresid.dat we provided.
Thus, we can obtain snapshot numbers for many probe/chain combinations as shown below:
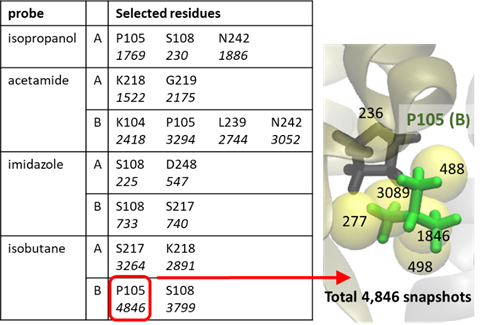
Figure 6. Probe molecules in a region of interest. Probe-specific high affinity residues and frequency of specific probe-amino acid contacts listed in the table, and illustrated for one specific case, isobutane near P105(B), in the diagram on the right. The frequency of contacts (listed in italic in the table) represents the total count of contacts observed in 10,000 frames from a 40 ns run. For example, in the case of P105(B) a total of 4,846 snapshots with one or more contacts with isobutane, distributed over 6 hot spots (see the corresponding counts shown for each hot spot), were observed.
3.5. Pharmacophore model from a snapshot with multiple probes
In the pharmmaker/CORE directory, there is a file called multipleprobe.sh for finding snapshots with multiple probes occupying hot spots near high affinity residues, which can be used as pharmacophore models. This file takes the outputs from the previous steps and can be run without any arguments from the command line as follows:
[outputs] bash ../pharmmaker/CORE/multiprobe.sh
Two output files are generated.
One output file, multipleprobe1.dat, is as below:
=========== multipleprobe1.dat ===========
2391 15
2462 14
2399 14
761 13
~~
=========== multipleprobe1.dat ===========
The other output file, multipleprobe2.dat, is as below:
=========== multipleprobe2.dat ===========
2391 out-A-105-IPRO-1.dat IPRO 7
2391 out-A-108-IMID-1.dat IMID 2
~~
=========== multipleprobe2.dat ===========
The first column is the frame number, the second column is the name of the file which is obtained from step 3.4, the third column is probe ID, and the fourth column is the probe residue number. Checking which residues and hot spots are involved with the probes at frame 2391 yields Fig 7.
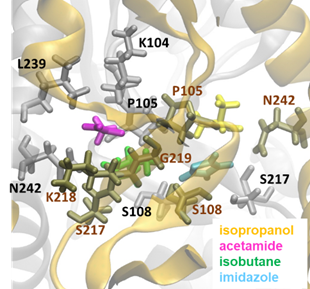
Figure 7. A snapshot from druggability simulations used for pharmacophore modeling. We observe an isopropanol close to P105(A) and N242(A), an acetamide close to K218(A), G219(A), K104(B), P105(B), L239(B), and N242(B), an imidazole close to S108(A) and S108(B) and S217(B), and an isobutane close to S217(A), K218(A), P105(B), and S108(B). Residues of subunit A are labelled in brown, and those of chain B, in black.