Trajectory Analysis II¶
This example shows how to perform a more elaborate calculations simultaneously. Radius of gyration, distance, psi angle calculated will be calculated using trajectory frames.
Setup environment¶
We start by importing everything from ProDy:
In [1]: from prody import *
In [2]: from pylab import *
In [3]: ion()
Parse structure¶
The PDB file provided with this example contains and X-ray structure which will be useful in a number of places, so let’s start with parsing this file first:
In [4]: structure = parsePDB('mdm2.pdb')
In [5]: structure
Out[5]: <AtomGroup: mdm2 (1449 atoms)>
This function returned a AtomGroup instance that stores all atomic
data parsed from the PDB file.
Handling multiple files¶
Trajectory is designed for handling multiple trajectory files:
In [6]: traj = Trajectory('mdm2.dcd')
In [7]: traj
Out[7]: <Trajectory: mdm2 (next 0 of 500 frames; 1449 atoms)>
In [8]: traj.addFile('mdm2sim2.dcd')
In [9]: traj
Out[9]: <Trajectory: mdm2 (2 files; next 0 of 1000 frames; 1449 atoms)>
Link trajectory to atoms¶
Atoms can be linked to the trajectory as follows:
In [10]: traj.link(structure)
In [11]: traj.setCoords(structure)
When an atom group is linked to a trajectory, frame coordinates parsed from trajectory files will overwrite coordinates of the atom group. By making atom selections, you can calculate and analyze different properties.
Setup for calculations¶
Let’s make atom selections for different types of calculations:
End-to-end distance¶
We select atoms from terminal residues and make an empty array whose length equal to the number of frames:
In [12]: nter = structure.select('name CA and resnum 25')
In [13]: cter = structure.select('name CA and resnum 109')
In [14]: e2e = zeros(traj.numFrames())
Radius of gyration¶
We select atoms protein atoms this calculation and make an empty array:
In [15]: protein = structure.select('noh and protein')
In [16]: rgyr = zeros(traj.numFrames())
A psi angle¶
We select a residue an make an empty array:
In [17]: res30 = structure['PPP', 'P', 30]
In [18]: res30
Out[18]: <Residue: PRO 30 from Chain P from mdm2 (14 atoms)>
In [19]: res30psi = zeros(traj.numFrames())
Perform calculations¶
We perform all calculations simultaneously as follows:
In [20]: for i, frame in enumerate(traj):
....: e2e[i] = calcDistance(nter, cter)
....: res30psi[i] = calcPsi(res30)
....: rgyr[i] = calcGyradius(protein)
....:
Let’s print part of results:
In [21]: e2e[:10]
Out[21]:
array([11.78980637, 14.12566566, 15.6633606 , 14.52022934, 16.45702362,
17.20821953, 16.45432854, 14.28651619, 11.59599113, 12.66241741])
In [22]: rgyr[:10]
Out[22]:
array([12.85520891, 12.98176002, 12.82791643, 12.91550011, 12.87289194,
12.92314255, 12.76350405, 12.85890813, 12.81869193, 12.76178599])
In [23]: res30psi[:10]
Out[23]:
array([149.81183155, 170.65785032, 139.9378317 , 156.36605157,
139.49376043, 151.11160105, 147.68076198, 151.81761523,
143.4179355 , 155.13133287])
Plot results¶
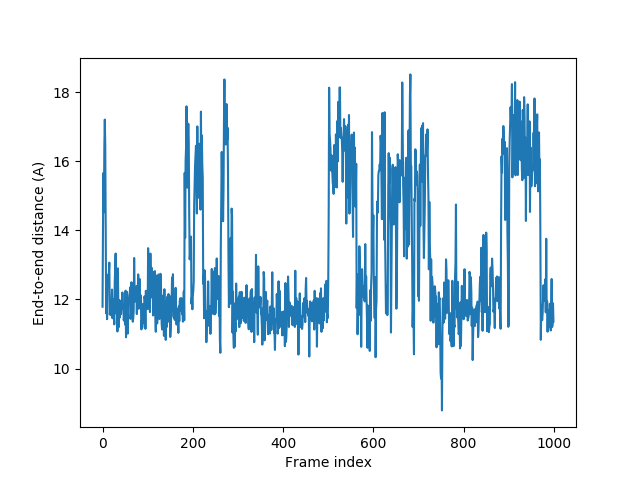
End-to-end distance¶
In [24]: plot(e2e);
In [25]: xlabel('Frame index');
In [26]: ylabel('End-to-end distance (A)');
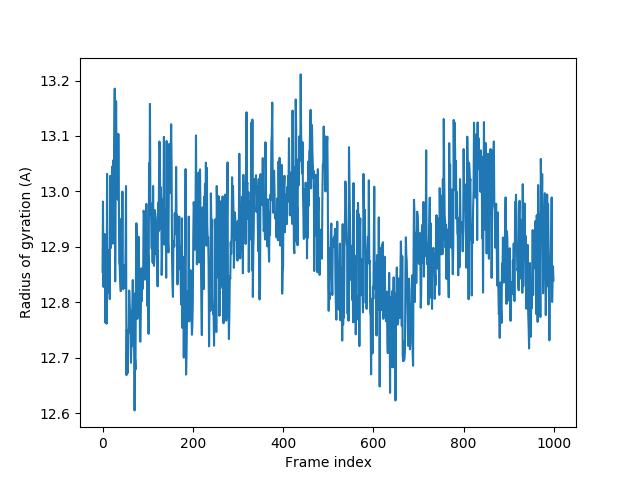
Radius of gyration¶
In [27]: plot(rgyr);
In [28]: xlabel('Frame index');
In [29]: ylabel('Radius of gyration (A)');
A psi angle¶
In [30]: plot(res30psi);
In [31]: xlabel('Frame index');
In [32]: ylabel('Residue 30 psi angle');
