Simulation Setup¶
A system that contains protein structures with water and counter ions in collective molecular dynamics simulations can be prepared using the following interface:
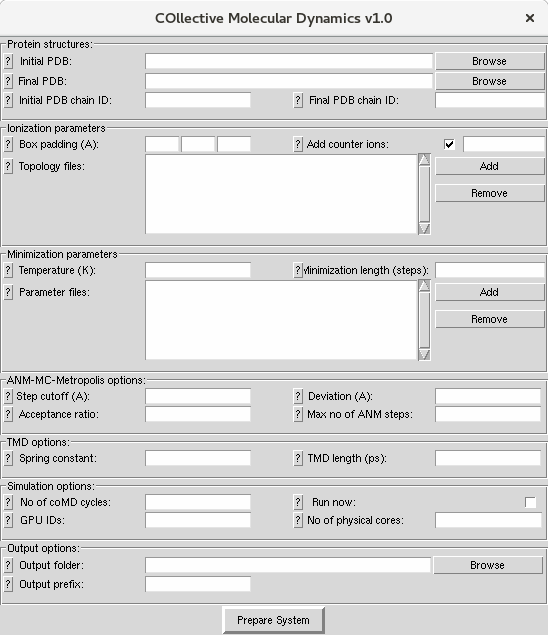
Input Files¶
.pdb files for both initial and final structure of the protein are required from the user. You can learn how to prepare these files from NAMD tutorials.
Protein Structures¶
- First, select initial
.pdb(structure) files and final.pdb(structure) files. You can use the adelynate kinase files provided for this tutorial (1ake.pdb, 4ake.pdb). Alternatively, structure and coordinate files for a protein of interest can also be used. These files should contain all atoms required for protein stability and function, these may include cofactors and metal atoms. Crystallographic water molecules may also be retained in the structure. - Select the corresponding chain IDs for initial and final structure. For adenylate kinase both chain IDs should be ‘A’. You can also select chains containing non-protein components such as membrane. If you leave this box blank, all protein atoms will be selected.
Ionization Parameters¶
- To build the topology of the structure, a topology file needs to be provided. This file will parametrize the topology of structures e.g. bond lengths and bond angles. If not given, it will use CHARMM36 all-hydrogen topology file for proteins and lipids. For more information visit Topology pages.
- Enter simulation box padding (distance from the protein to box surfaces) in x, y and z and select whether you would like to add counter ions and what concentration. Collective Molecular Dynamics GUI uses Solvate and Autoionize plugins to add water and ions.
Minimization Parameters¶
- For every cycle, the structures (protein and water) will be minimized by using NAMD. For parametrization of molecular dynamics simulation in NAMD, an input file is required. If not given, it will use CHARMM36 all-hydrogen parameter files. For more information visit Parameter pages.
- For minimization, two additional parameters are required. The first one is the temperature at which the minimization will be performed and the second one is the total number of steps to perform minimization.
Conformational Sampling with ANM Modes¶
For sampling of conformations with ANM modes, there are some optional parameters that all take default (recommended) values.
ANM cutoff is the parameter to determine the contacts on the proteins for ANM calculations (default 15).
The second parameter is the maximum deviation for each step - this would be the RMSD for the first (largest amplitude) mode. From this, we derive a scale factor for perturbing structure in the direction of a given ANM mode. The default is 2 A.
Other important parameters are the acceptance ratio (default 0.9), which determines how many wrong direction moves are accepted versus right direction ones, and the maximum number of steps (default 100).
Targeted Molecular Dynamics Parameters¶
For every cycle, after conformational sampling with ANM modes towards to the other structure, the side chains of the protein need to move by performing a fast targeted molecular dynamics (TMD) simulation. In TMD simulations, two parameters are required. The first one is the spring constant (kcal/mol/A^2) and the second one is the length of TMD simulation (ps). For further information visit TMD pages.
Simulation Options¶
coMD simulations will run in cycles and the number of cycles is the global parameter for them. The simulation will run until the number of cycles is achieved or the RMSD between initial and final structure is less than 1.5 A or the reduction in the RMSD is less than 0.15 A. Also provided in this block are which GPUs and the number of physical cores to use for parallel NAMD simulations, and the option to run now or not. You would not run now if you intended to copy the files to a cluster for example.
Output Options¶
Here you select an output folder and a file prefix to distinguish different runs. Performing multiple simulations to see whether results are reproducible is always a good idea.
Output Files¶
There will be an output folder with a python script (.py file), a tcl script (.tcl file), and two .pdb and .psf files. The tcl script should run from vmd.
For a summary of contents of the final system, see prefix.log file.
Simulation¶
Now you need to run coMD simulations. To perform those simulations it is possible to use the following command:
vmd –dispdev text –e your_output_prefix.tcl
If you want to run this simulation on a cluster, copy this directory on cluster and put the command on the queue system.
When simulations are complete, you can continue with following analysis steps.