Visualization & Analysis¶
Following interface can be used to generate a quick visualization of results from druggability analysis.
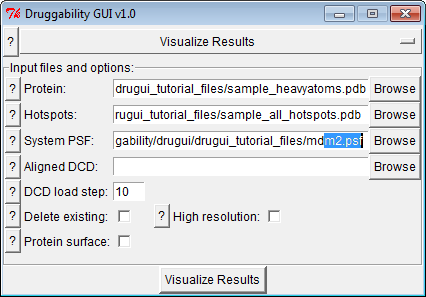
Input Files¶
Input files are prefix_heavyatoms.pdb and other PDB files
in prefix folder.
Options & Parameters¶
- If you have outputted aligned trajectory in grid calculation step, you can select to load it too.
- Optionally, molecules present in VMD can be deleted, high resolution representations and a protein surface representation can be generated.
Probe binding spots¶
When results are loaded, you will see a representation similar to the following:
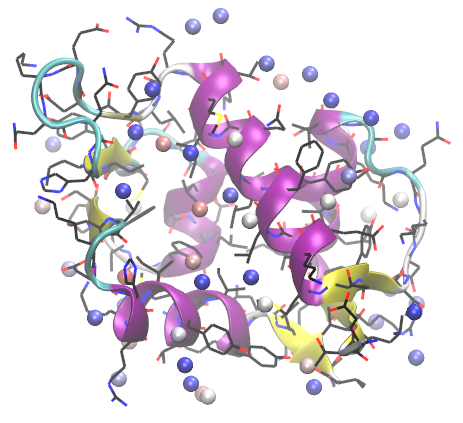
Each sphere corresponds to a probe binding spot. Spheres are colored according to their binding free energies. Red most sphere has the lowers binding free energy.
Binding free energies of probes can be found in the logfile:
defaults is initialized.
defaults working directory is set to "defaults".
Druggability Analysis defaults is initialized.
Parameter: temperature 300.00 K
Parameter: delta_g -1.000 kcal/mol
Parameter: n_probes 7
Parameter: min_n_probes 6
Parameter: merge_radius 5.5 A
Parameter: low_affinity 10.00 uM
Parameter: n_solutions 3
Parameter: max_charge 2.0 e
Parameter: n_charged 3
Parameter: n_frames 1
Parsing OpenDX file defaults_IPRO.dx.
defaults_IPRO was parsed in 0.45s
Parsing OpenDX file defaults_IPAM.dx.
defaults_IPAM was parsed in 0.45s
Parsing OpenDX file defaults_ACAM.dx.
defaults_ACAM was parsed in 0.45s
Parsing OpenDX file defaults_ACET.dx.
defaults_ACET was parsed in 0.45s
Searching probe binding hotspots with deltaG less than -1.00 kcal/mol (~5 folds enrichment).
51 all-probes binding spots were identified in 1.07s.
Minimum binding free energy is -2.73 kcal/mol.
Hotspot 1 -2.73 kcal/mol 100.0% IPRO
Hotspot 2 -2.36 kcal/mol 100.0% IPRO
Hotspot 3 -2.34 kcal/mol 95.3% ACET 2.5% IPRO 2.2% ACAM
Hotspot 4 -2.33 kcal/mol 100.0% IPRO
Hotspot 5 -2.28 kcal/mol 93.4% IPRO 3.8% ACAM 2.8% IPAM
Hotspot 6 -2.23 kcal/mol 98.8% IPRO 1.2% ACAM
Hotspot 7 -2.19 kcal/mol 94.6% IPRO 2.9% ACAM 2.5% ACET
Hotspot 8 -2.13 kcal/mol 99.5% IPRO 0.3% ACET 0.2% ACAM
Hotspot 9 -2.13 kcal/mol 96.9% IPRO 3.1% ACAM
Hotspot 10 -2.02 kcal/mol 84.5% IPRO 15.5% ACAM
Hotspot 11 -2.00 kcal/mol 83.9% IPAM 12.7% IPRO 3.4% ACAM
Hotspot 12 -1.98 kcal/mol 93.2% IPRO 4.7% ACAM 2.1% IPAM
Hotspot 13 -1.91 kcal/mol 97.2% IPRO 2.8% ACAM
Hotspot 14 -1.89 kcal/mol 80.7% IPRO 18.0% ACET 1.3% ACAM
Hotspot 15 -1.85 kcal/mol 94.5% IPRO 4.7% IPAM 0.7% ACET 0.2% ACAM
Hotspot 16 -1.79 kcal/mol 82.5% IPRO 10.2% ACAM 5.6% ACET 1.6% IPAM
Hotspot 17 -1.75 kcal/mol 97.6% IPRO 2.4% ACAM
Hotspot 18 -1.72 kcal/mol 98.9% IPRO 0.6% ACAM 0.5% IPAM
Hotspot 19 -1.70 kcal/mol 46.3% ACET 45.4% IPRO 5.4% ACAM 2.9% IPAM
Hotspot 20 -1.56 kcal/mol 63.6% IPRO 22.7% ACET 11.7% ACAM 2.0% IPAM
Hotspot 21 -1.53 kcal/mol 79.7% ACET 20.3% IPRO
Hotspot 22 -1.44 kcal/mol 95.7% IPRO 4.3% ACAM
Hotspot 23 -1.40 kcal/mol 87.4% IPRO 11.0% IPAM 1.6% ACAM
Hotspot 24 -1.36 kcal/mol 50.2% ACET 49.8% IPRO
Hotspot 25 -1.30 kcal/mol 80.4% ACET 18.9% IPRO 0.7% ACAM
Hotspot 26 -1.30 kcal/mol 72.1% IPRO 16.0% ACET 6.6% IPAM 5.4% ACAM
Hotspot 27 -1.27 kcal/mol 54.5% IPRO 24.3% ACET 21.3% ACAM
Hotspot 28 -1.26 kcal/mol 88.4% ACET 9.0% IPRO 2.5% ACAM
Hotspot 29 -1.26 kcal/mol 78.4% ACET 18.6% IPRO 3.0% ACAM
Hotspot 30 -1.25 kcal/mol 89.8% IPRO 8.7% ACAM 1.5% IPAM
Hotspot 31 -1.24 kcal/mol 97.7% IPRO 2.3% ACAM
Hotspot 32 -1.23 kcal/mol 83.9% IPRO 16.1% ACAM
Hotspot 33 -1.23 kcal/mol 85.7% IPAM 13.5% IPRO 0.8% ACAM
Hotspot 34 -1.21 kcal/mol 94.0% IPRO 5.8% ACAM 0.3% IPAM
Hotspot 35 -1.20 kcal/mol 90.2% IPRO 6.4% IPAM 3.4% ACAM
Hotspot 36 -1.15 kcal/mol 90.3% IPRO 8.8% ACAM 0.9% ACET
Hotspot 37 -1.14 kcal/mol 88.9% IPRO 10.2% ACET 0.9% ACAM
Hotspot 38 -1.13 kcal/mol 94.7% IPRO 5.3% ACAM
Hotspot 39 -1.11 kcal/mol 99.0% ACET 0.6% IPRO 0.3% ACAM
Hotspot 40 -1.10 kcal/mol 97.4% IPRO 2.3% ACAM 0.3% IPAM
Hotspot 41 -1.09 kcal/mol 85.9% IPRO 8.7% ACAM 5.0% IPAM 0.3% ACET
Hotspot 42 -1.08 kcal/mol 100.0% IPRO
Hotspot 43 -1.08 kcal/mol 100.0% IPRO
Hotspot 44 -1.08 kcal/mol 96.6% IPRO 3.4% ACAM
Hotspot 45 -1.08 kcal/mol 55.8% IPAM 33.6% IPRO 10.6% ACAM
Hotspot 46 -1.06 kcal/mol 97.9% IPRO 2.1% ACAM
Hotspot 47 -1.06 kcal/mol 86.0% ACET 11.6% IPRO 2.1% ACAM 0.4% IPAM
Hotspot 48 -1.05 kcal/mol 96.8% IPRO 3.2% ACAM
Hotspot 49 -1.03 kcal/mol 100.0% IPRO
Hotspot 50 -1.01 kcal/mol 86.7% IPRO 11.8% IPAM 1.5% ACAM
Hotspot 51 -1.01 kcal/mol 85.6% IPRO 8.4% ACET 6.1% ACAM
IPRO: 39 isopropanol binding hotspots were identified.
IPRO: lowest binding free energy is -2.73 kcal/mol.
IPAM: 3 isopropylamine binding hotspots were identified.
IPAM: lowest binding free energy is -2.00 kcal/mol.
ACAM: 0 acetamide binding hotspots were identified.
ACET: 9 acetate binding hotspots were identified.
ACET: lowest binding free energy is -2.34 kcal/mol.
Clustering probe binding hotspots.
Clustering completed in 2.64ms.
1 potential sites are identified.
Calculating achievable affinity ranges.
Site 1: 16 probe binding hotspots
Site 1: Lowest probe binding free energy -2.36 kcal/mol
Site 1: Average probe binding free energy-1.56 kcal/mol
Site 1: Total of 70 solutions.
Achievable affinities for site 1
-log10(affinity)
#-------------#
9.53 |o |
9.28 |-o |
9.03 |--------o |
8.79 |----------o |
8.54 |-----------o |
8.29 |---------o |
8.05 |--------o |
7.80 |-----o |
7.56 |----o |
7.31 |----o |
#-------------#
0 5 10
Site 1: Lowest drug-like binding free energy -13.07 kcal/mol
Site 1: Highest drug-like affinity 0.298 nM
Site 1: Solution 1 binding free energy -13.07 kcal/mol
Site 1: Solution 1 affinity 0.298 nM
Site 1: Solution 1 total charge 0.02 e
Site 1: Solution 1 number of hotspots 7
Site 1: Solution 1 approximate volume 450.58 A^3
Site 1: Solution 2 binding free energy -12.66 kcal/mol
Site 1: Solution 2 affinity 0.593 nM
Site 1: Solution 2 total charge -0.03 e
Site 1: Solution 2 number of hotspots 7
Site 1: Solution 2 approximate volume 449.28 A^3
Site 1: Solution 3 binding free energy -12.49 kcal/mol
Site 1: Solution 3 affinity 0.780 nM
Site 1: Solution 3 total charge 0.03 e
Site 1: Solution 3 number of hotspots 7
Site 1: Solution 3 approximate volume 451.70 A^3
Hotspots are written into file defaults/defaults_all_hotspots.pdb.
defaults is cPickled into file defaults/defaults.dso.gz.
Logfile lists all probe binding spots, their binding free energies, and fractional contribution of different probe types to the hotspot.
Druggable Sites¶
Druggable sites are identified by clustering probe binding spots and merging them to identify subsets of binding spots that have a size similar to that of a drug-like molecule. After results load, you will see a list of molecules in VMD Main for each druggable site and solutions therein. You can toggle displayed molecules to see locations of different sites and solutions.
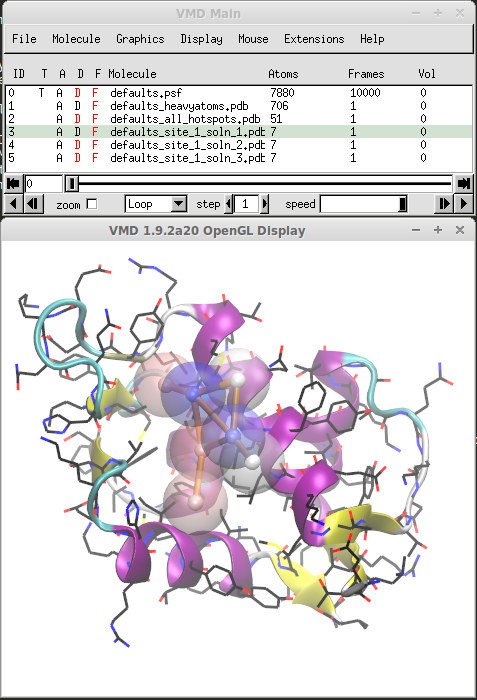
Figure shows the best solution for protein MDM2. The maximal achievable affinity (druggability index) for this solution is 0.3 nM or, in terms of free energy, it is -13 kcal/mol. You can find such information in the log file shown above.
Probe binding hotspots and protein structure shown above can be found among tutorial files. These results of course deserve a more detailed analysis, and some things that can be done include:
- looking into types of probes that contribute to a given binding spot and types of amino acid residue interacting with the binding spot
- visualizing trajectories (in which probes are wrapped) to see specific interactions and residence time of probes at a given binding spot
- comparing results from simulations in presence and absence of probes to see how binding site shape is affected by ligand binding
- looking into other structures of the target protein (ligand binding sites, crystal contacts, protein interfaces) to see whether observations in simulations are supported by interactions determined experimentally
Visualize Probe Grids¶
Finally, you can visualize probe occupancy grids using VMD. Simply load .dx
files and create isovolume representations. An example
is shown below for sample_IPRO.dx, and other grid files.
Mesh surfaces correspond to locations that were highly enriched with probes. Coloring
is as follows:
- isopropanol: green (high enrichment at the binding site)
- acetate: red (enrichment at the surface, not that proetein has +5 net charge)
- isopropylamine: blue (few interaction spots)
- acetamide: magenta (not observed to interact with this protein much)
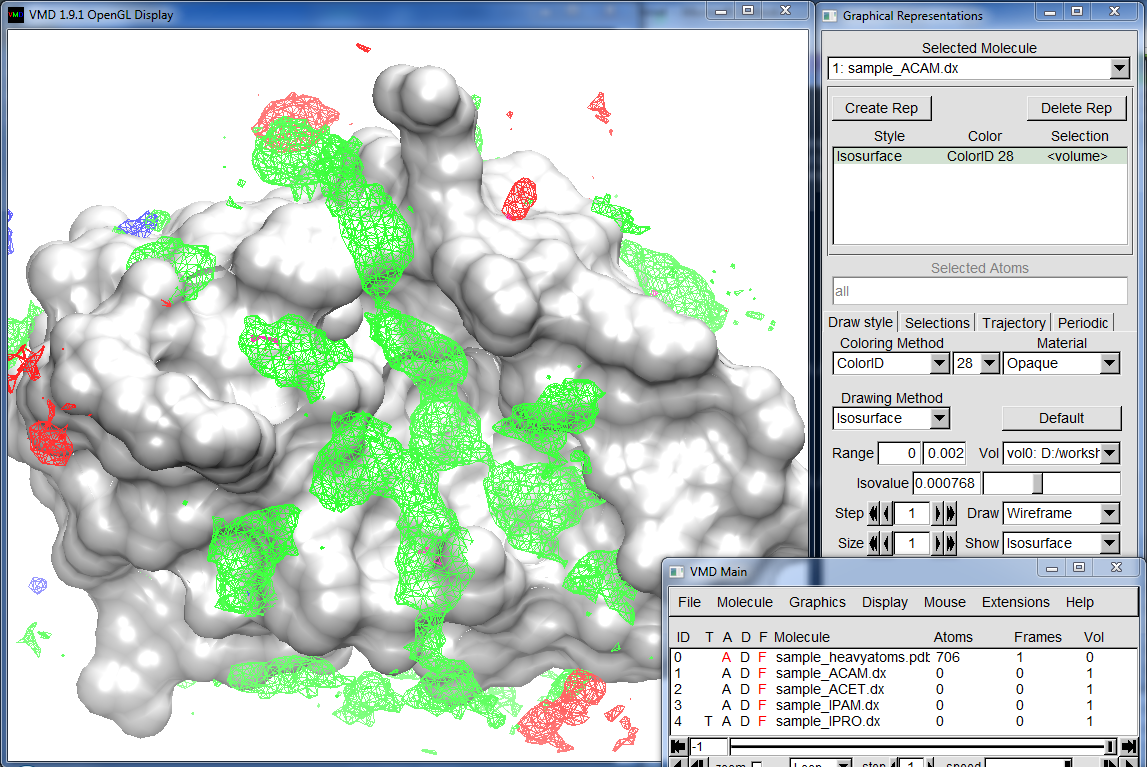
Note that values in occupancy grids is the count of central carbon atoms of probe molecules. Since the grid elements (voxels) are small (0.5A dimension), the occupancy numbers are small. You will need to adjust Isovalue value in VMD Representations window to make grid elements visible.
Similar representations can be generated for water or other atom type specific grids too.