
Dynamics of p38 MAP kinase inferred from a structural ensemble using PCA is compared to intrinsic dynamics of the protein modeled using ANM. See PCA of X-ray structures or Bioinformatics article for more details.
Dynamics of p38 MAP kinase inferred from a structural ensemble using PCA is compared to intrinsic dynamics of the protein modeled using ANM. See PCA of X-ray structures or Bioinformatics article for more details.
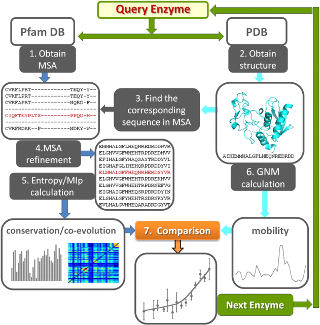
Workflow for comparative analysis of sequence evolution and structural dynamics is shown. See Evol Applications or Mol Biol Evol article for more details.
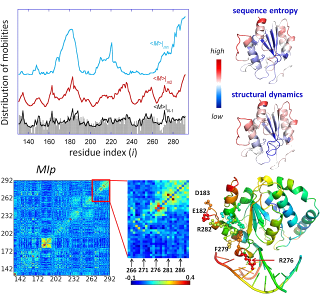
Results from comparative analysis of residue conservation, conformational mobility, and coevolutionary patterns for uracil-DNA glycosylase. See Mol Biol Evol article or Conservation and Coevolution Analysis for more details.
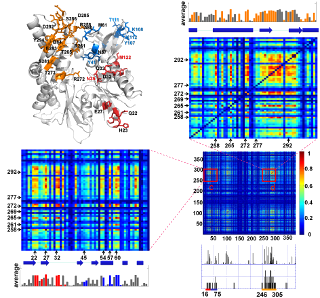
Coevolution of NEF-binding residues analzyed using mutual information is displayed for the Hsp70 ATPase domain. See PLoS Comp Biol article or Conservation and Coevolution Analysis for details.
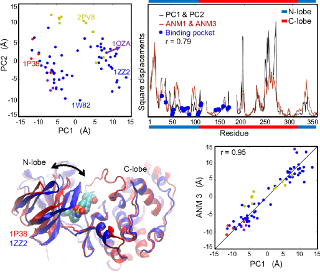
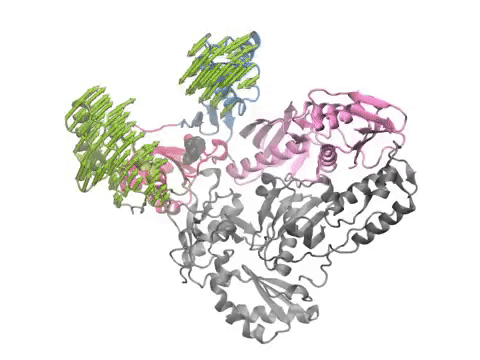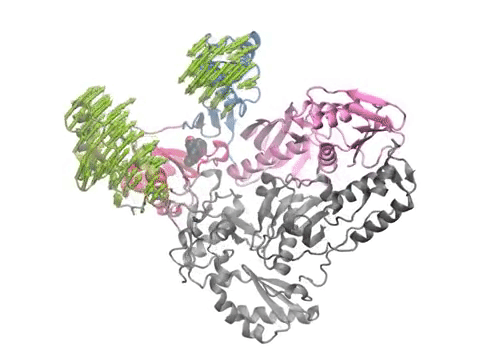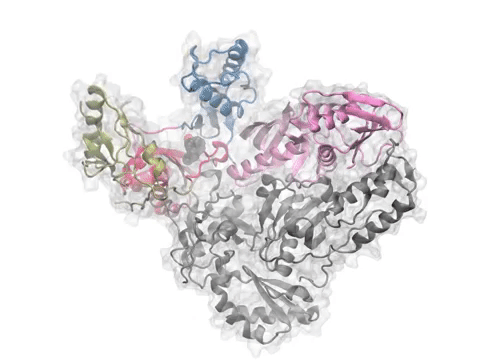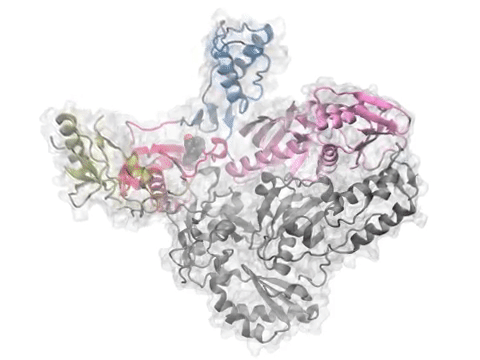
Comparative analysis of p38 MAP kinase dynamics from experiments (PCA) and theory (ANM). See the PNAS article or figure for details.
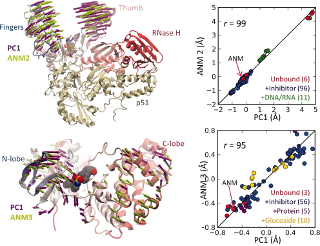
Comparative analysis of dynamics of drug target proteins and model systems from experiments (PCA) and theory (ANM). See the Protein Science article for details.
Comparative analysis of p38 MAP kinase dynamics from experiments (PCA), simulations (EDA), and theory (ANM). See the Protein Science article for details.
Animation shows HIV-1 reverse transcriptase functional motions calculated using anisotropic network model. Arrows and animations are generated using NMWiz VMD plugin. See NMWiz tutorial for usage examples.

You can make a quick protein representation in interactive sessions using showProtein() function.

NMWiz is designed for picturing normal modes easy. Image shows arrows from slowest three ANM modes for p38 MAP kinase centered at the origin. They indeed align with planes normal to each other.
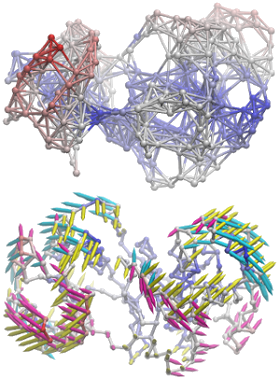
NMWiz makes depicting elastic network models and protein motions predicted with them easy. Image shows ANM model for p38 MAP kinase and three slow ANM modes (below).
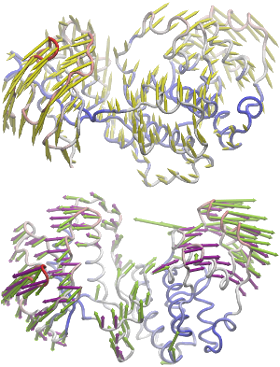
NMWiz can be used to comparative dynamics inferred from experimental datasets and predicted using theory.
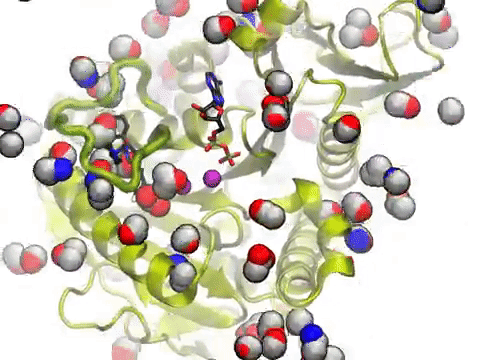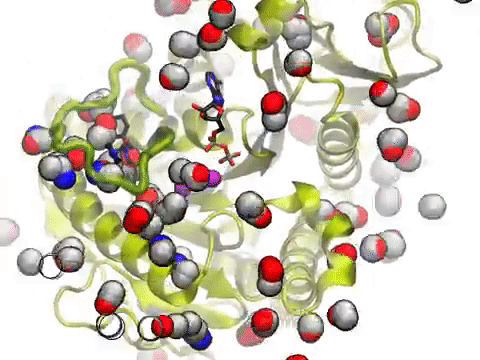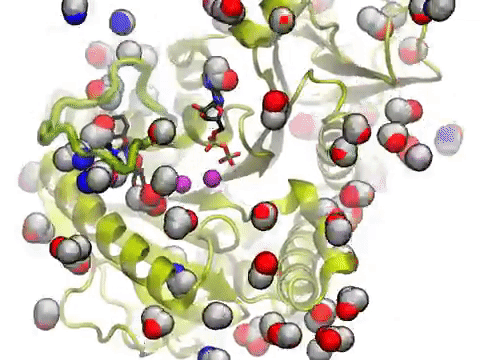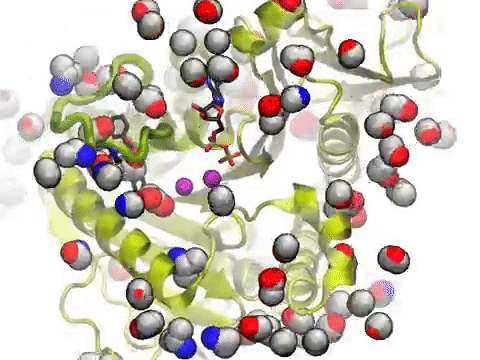
The movie shows a molecular dynamics simulation for assessing the druggability of kinesin eg5. NMWiz VMD plugin. See NMWiz tutorial for usage examples.
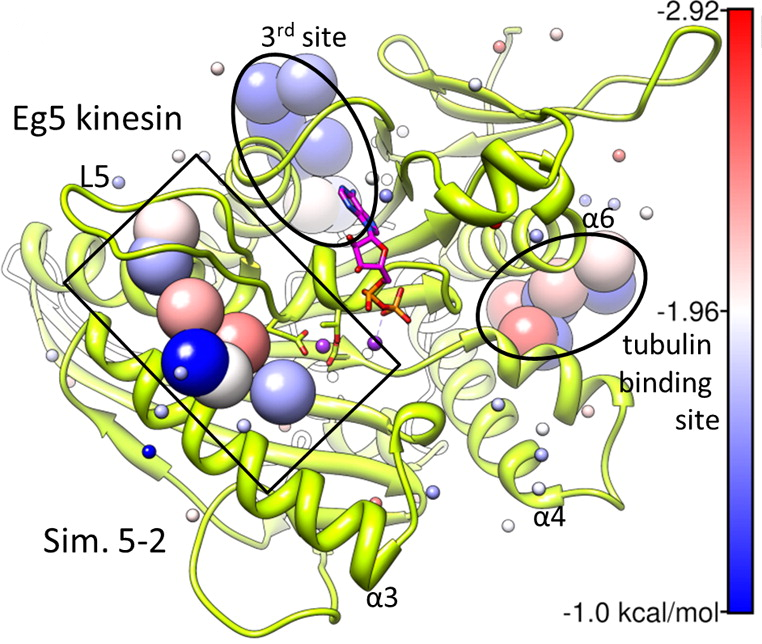
Kinesin Eg5 druggable sites, including allosteric inhibitor binding site and and tubulin binding site, identified by simulations are shown. See our publication for details.
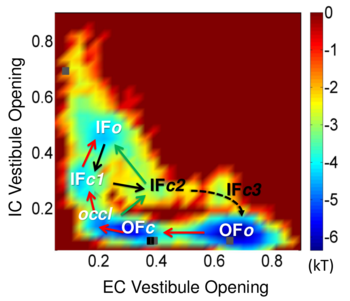
Sampling of the functional substates (inward-facing (IF) or outward-facing (OF), in closed (c) or open (o) forms) of LeuT using coMD simulations. See publication for details.
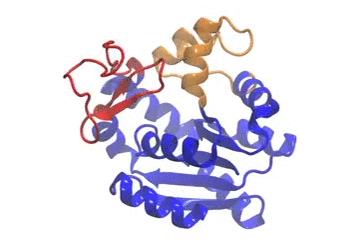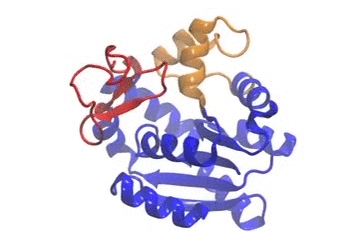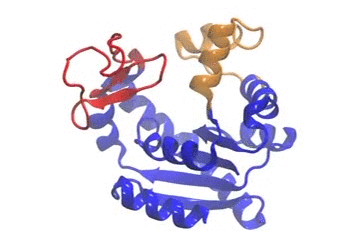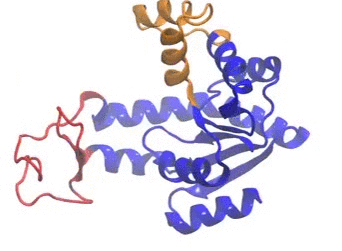
The movie illustrates a coMD trajectory for adenylate kinase. NMWiz VMD plugin. See NMWiz tutorial for usage examples.
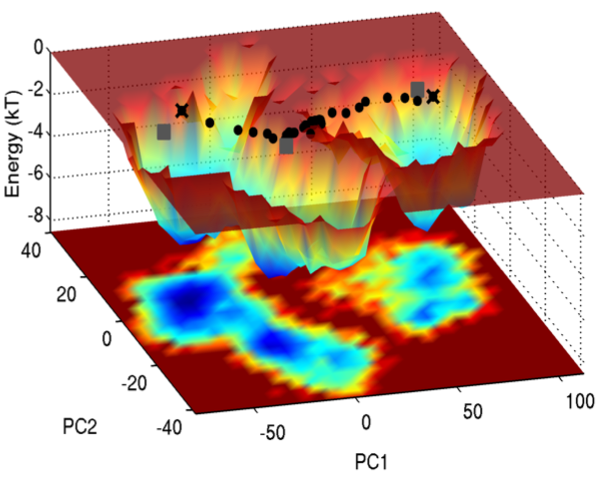
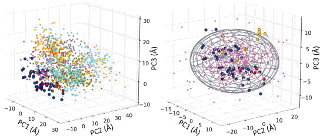
Energy landscape in the space of principal coordinates.
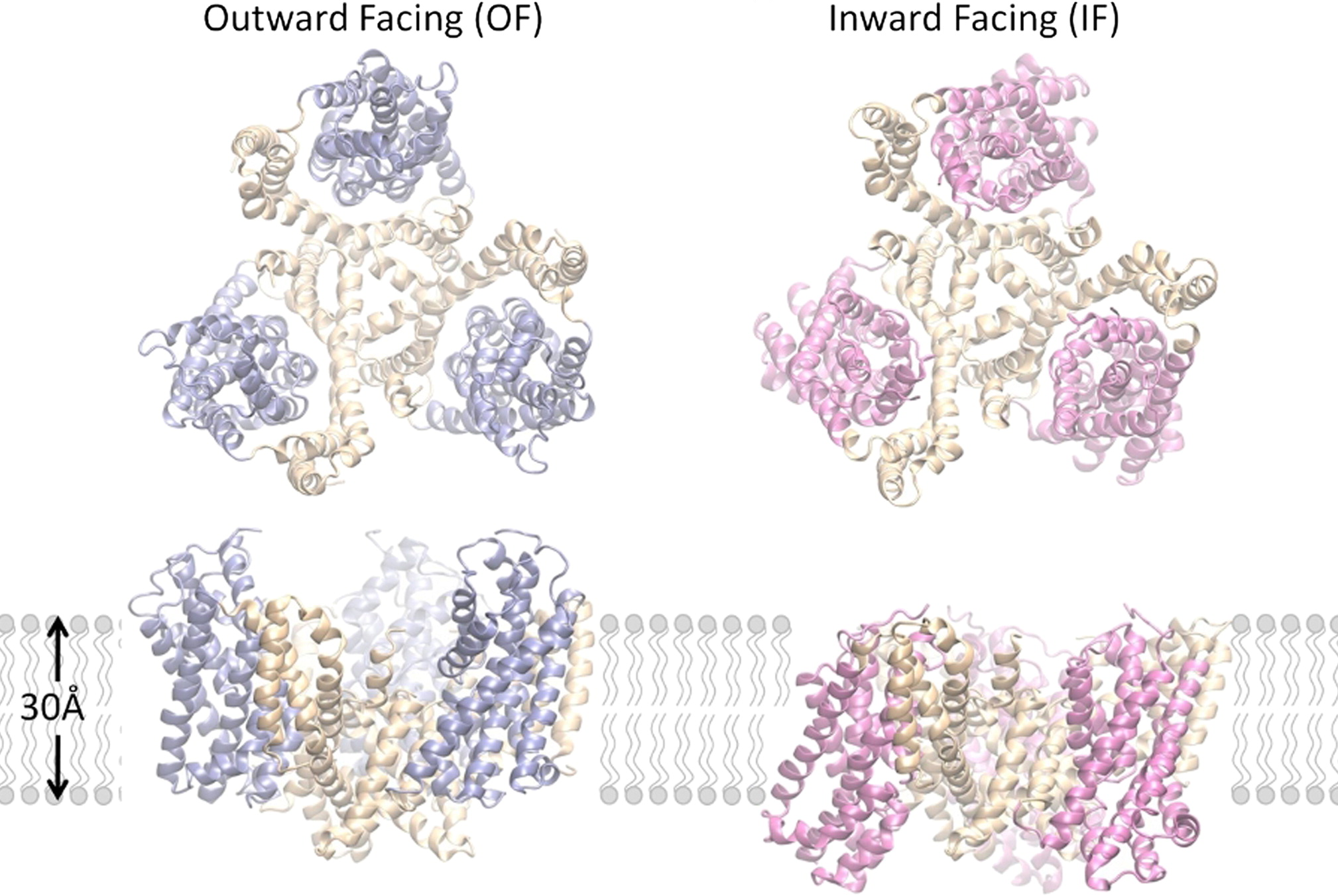
Outward-facing (OF) and inward-facing (IF) structures of GltPh show a large displacement of the core domains. See publication for details.
The second mode of the OF structure moves all three transport domains simultaneously through the membrane in a ‘lift-like’ motion. See publication for details.
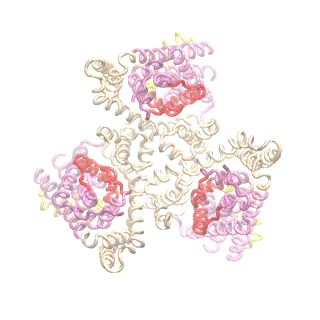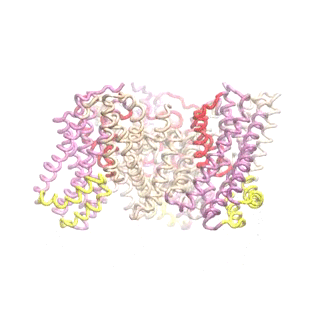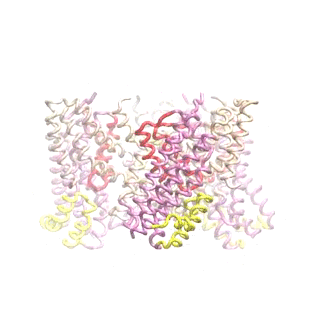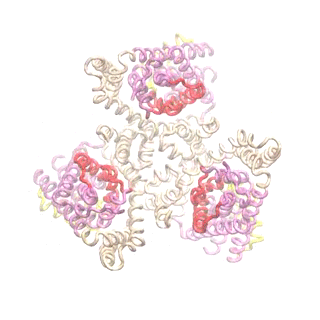
The second mode of the IF structure moves all three transport domains simultaneously through the membrane in a ‘lift-like’ motion. See publication for details.
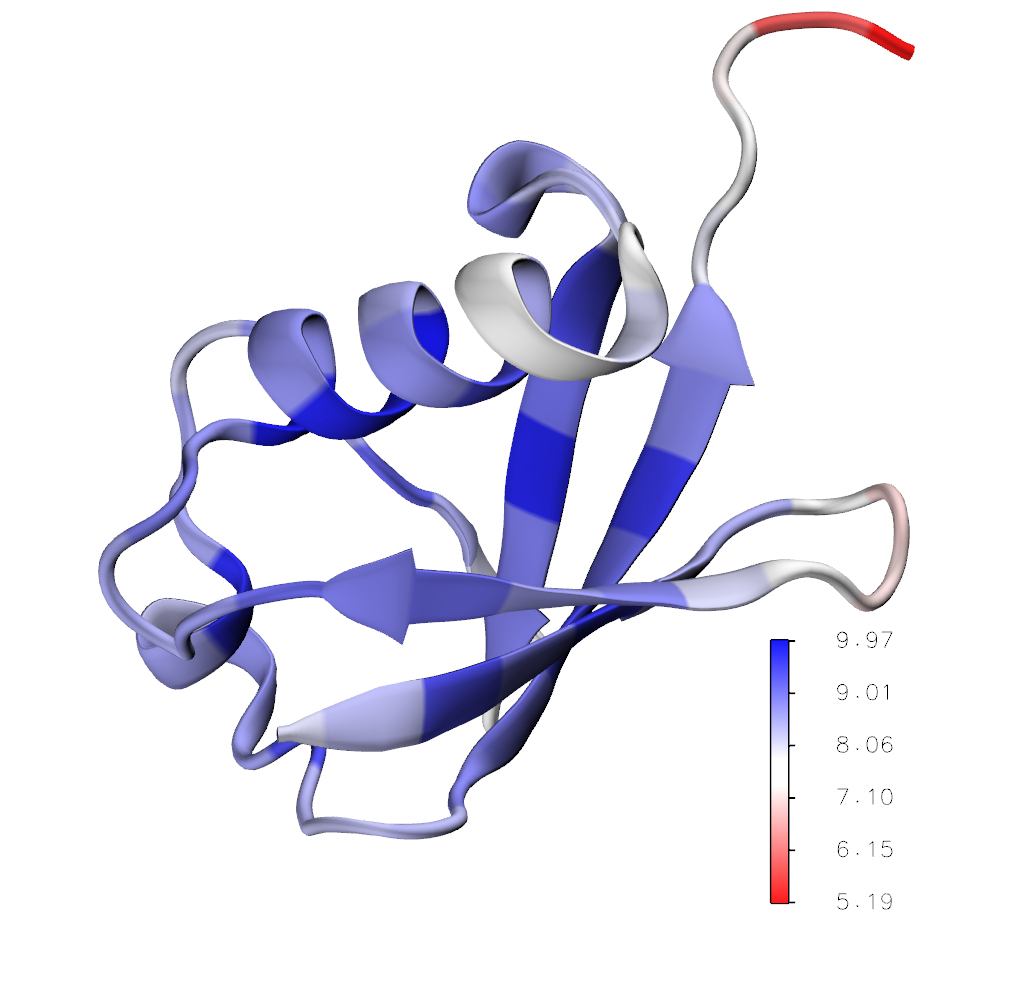
Deformability profile of ubiquitin (PDB code: 1UBI). Structure is automatically uploaded to VMD program where blue color shows regions which are mechanically more resistant to the external force.
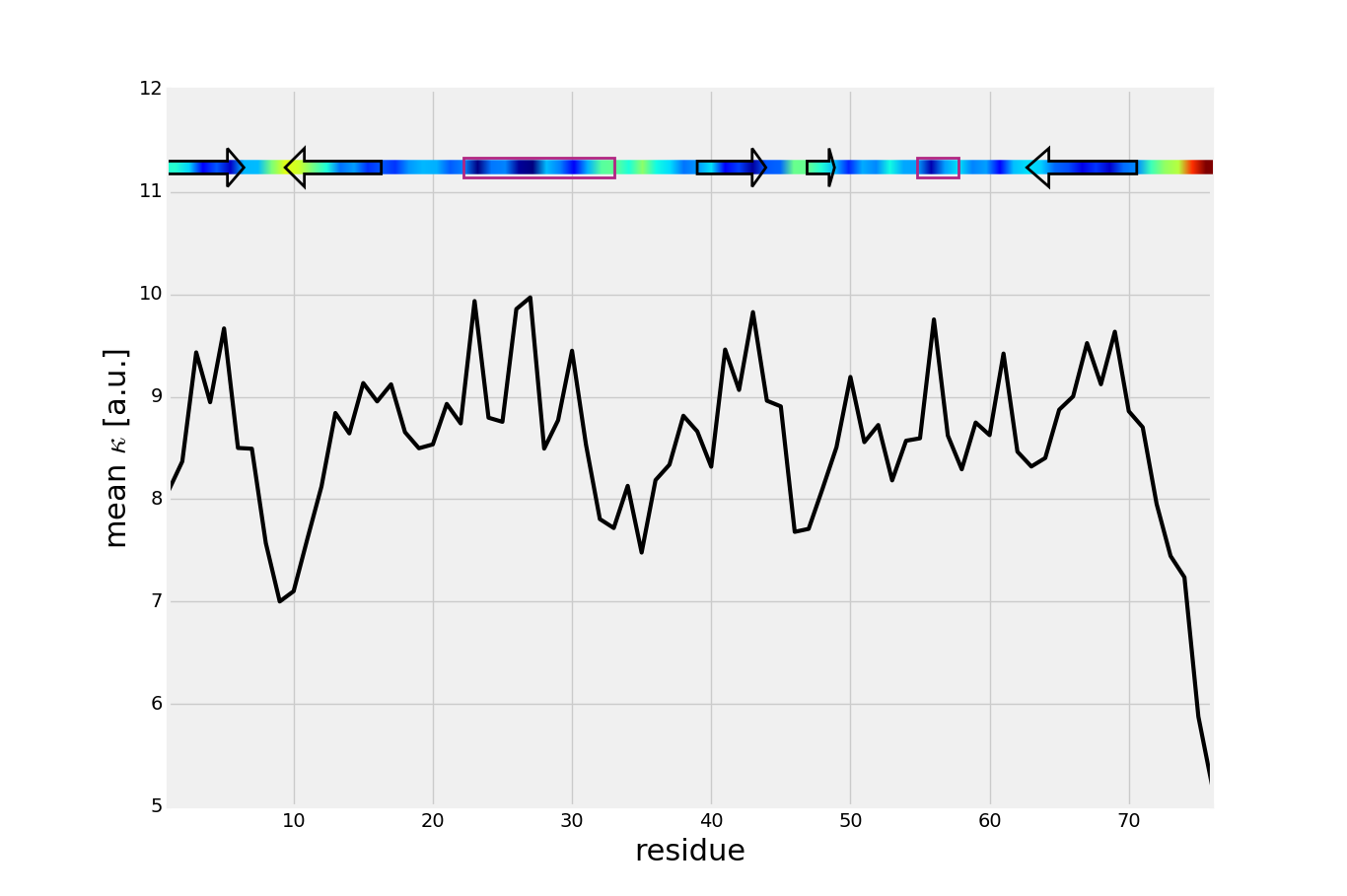
Mean value of effective spring constant (calculated from mechanical stiffness matrix) with secondary structure of ubiquitin. Blue color indicates mechanically strong regions.
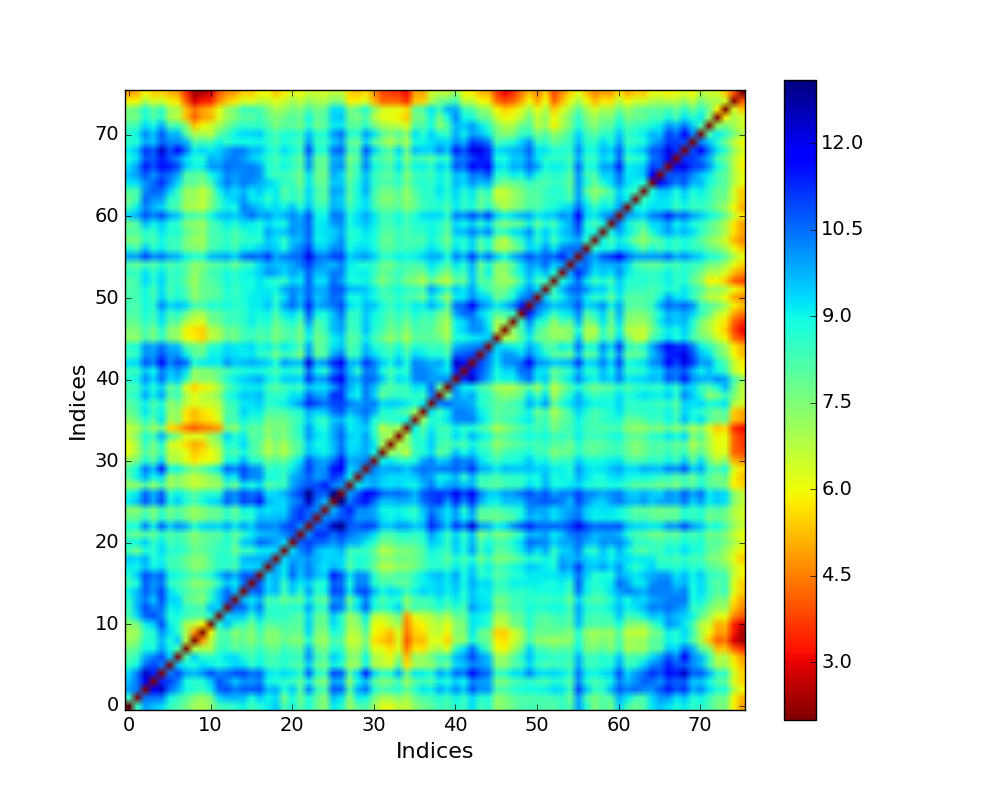
Mechanical Stiffness Map with effective force constant in a color bar (blue - strong regions, red - weak regions) for ubiquitin.

Workflow for GNM analysis of chromatin dynamics. See publication for details.

Covariance matrix of chromosome 17 of human B cells. Structural domains and CCDDs are identified and outlined. See publication for details.

3D Laplacian embedding of chromosome 17 loci using the first three principal modes. See ChromD tutorial for details.
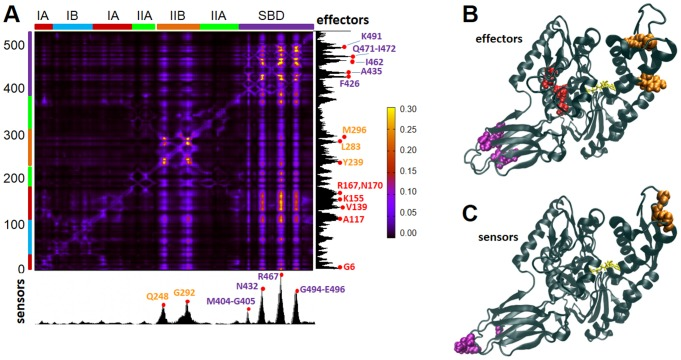
Perturbation response scanning of the Hsp70 chaperone reveals interdomain allostery. See publication for details.
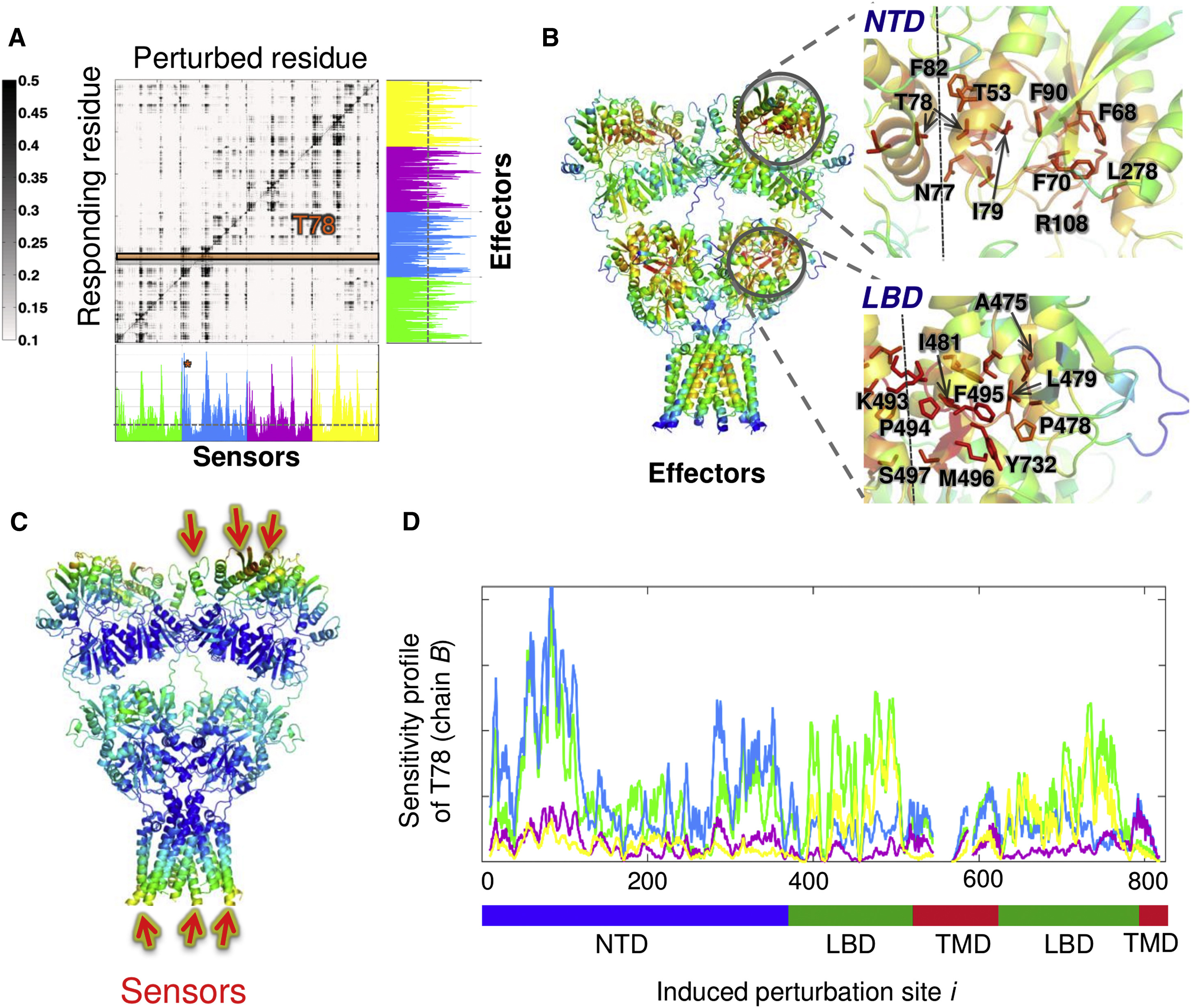
Perturbation response scanning of the AMPA-type glutamate receptor reveals sensors and effectors for allosteric signaling. See publication for details.
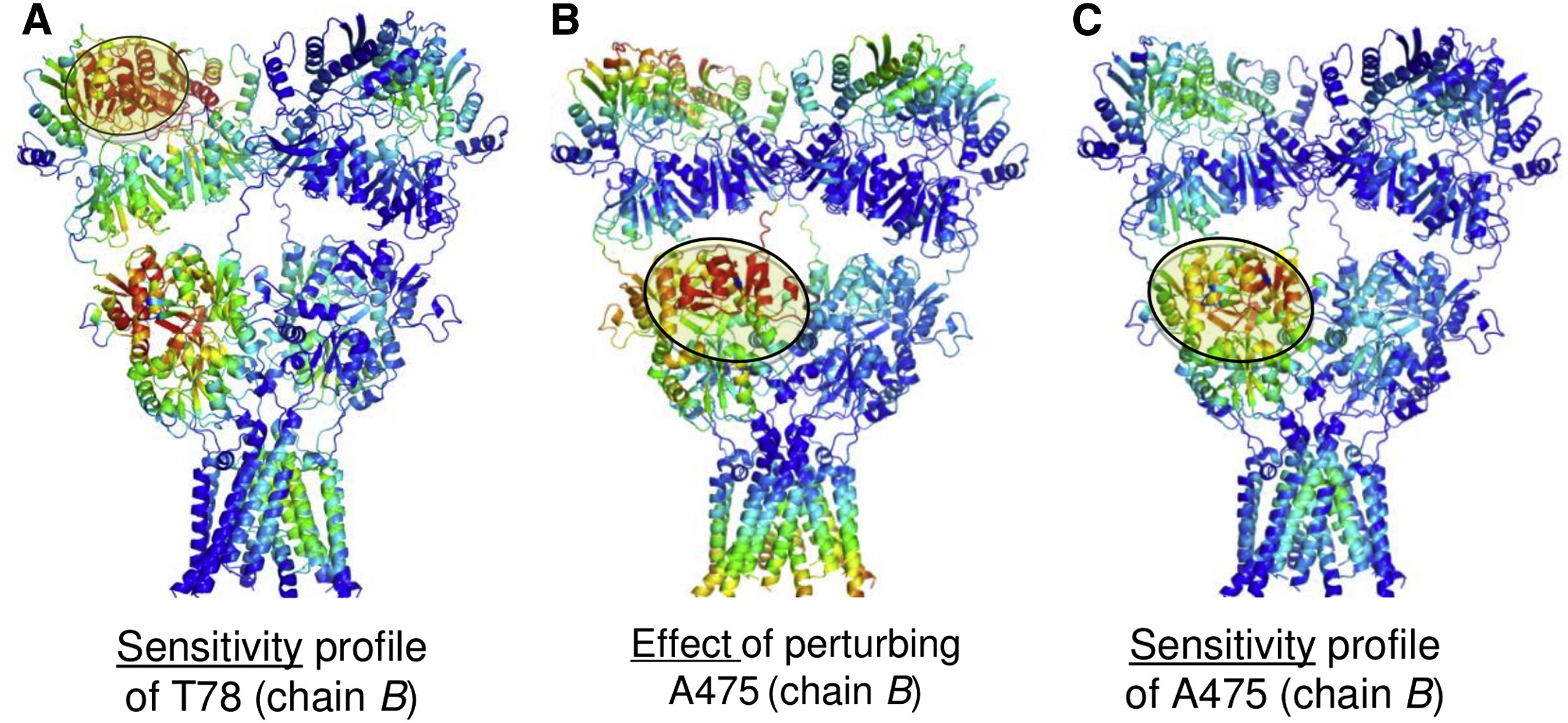
A more in-depth analysis of the PRS matrix reveals interdomain signaling in the AMPA receptor. See publication for details.
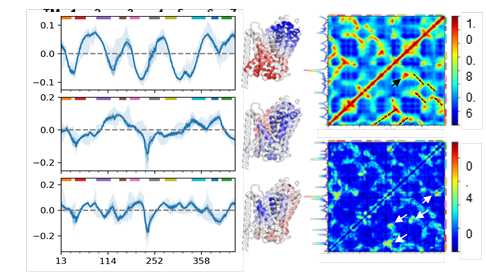
The left panels show the three softest GNM modes (blue lines) and their standard deviations (faint blue bands). Red and blue regions in the corresponding ribbon diagrams show regions moving in opposite directions. The right panel has the average cross-correlation matrix from the first 20 global modes (top) and its standard deviation (bottom).
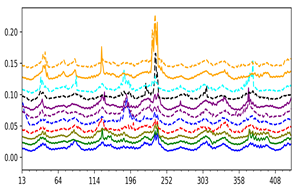
Square fluctuations calculated from the top 5 global modes are shown for a number of LeuT fold family members, revealing similarities and subfamily- or conformation-dependent differences.
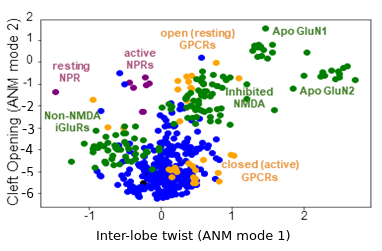
Type-I periplasmic binding protein domains are mapped onto the first two signature ANM modes. These domains, found in a range of proteins including bacterial solute carriers and eukaryotic receptors, have two lobes that undergo well-characterised conserved motions that are evident from comparison of structures. SignDy reveals such conserved dynamics.
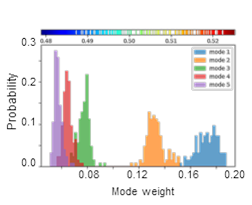
SignDy analysis allows a comparison of the frequency dispersion of family members. The distribution of inverse eigenvalues is shown for the softest five modes for TIM barrel fold family.
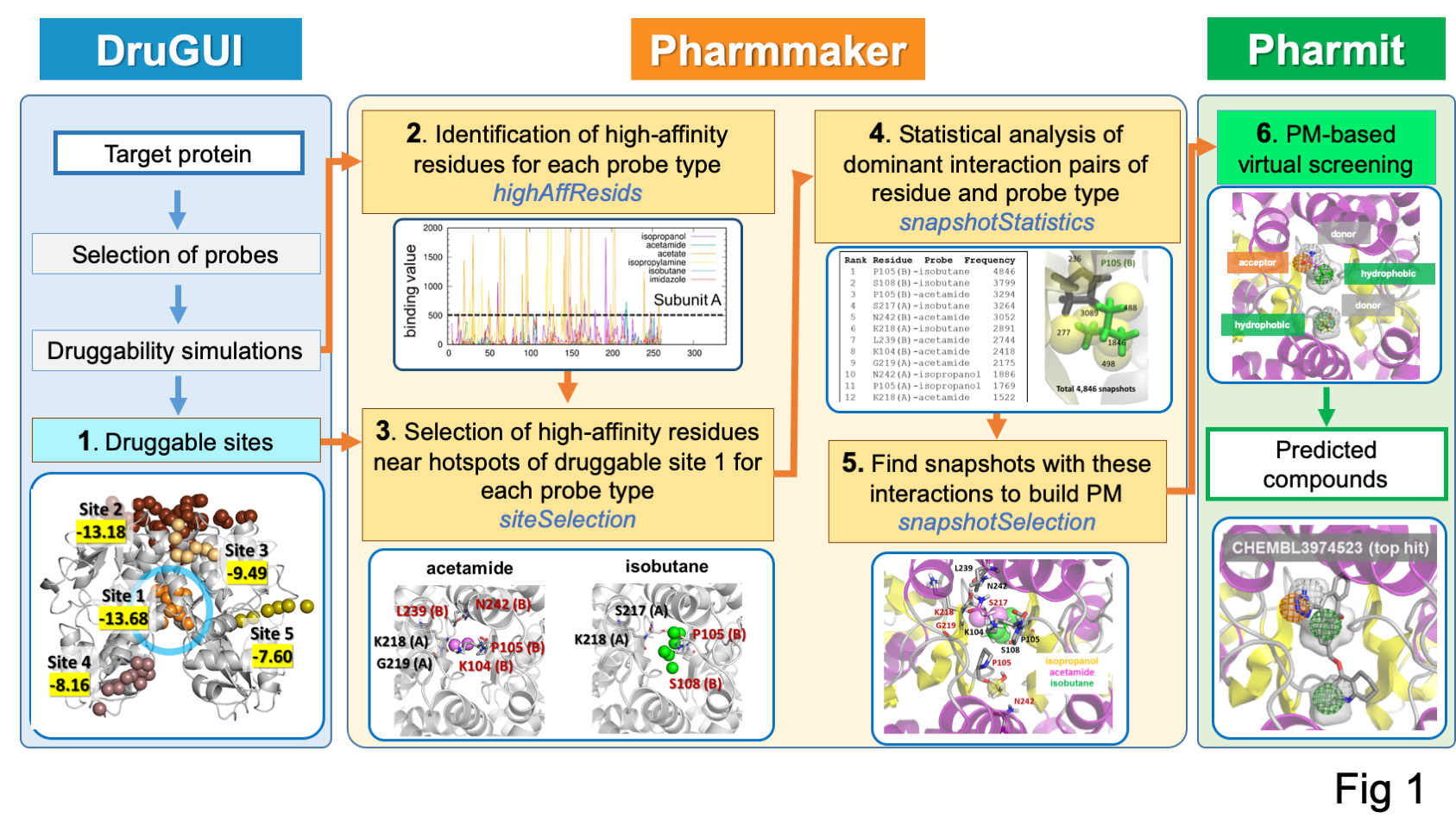
Pharmmaker (center) includes four main programs (steps 2 to 5 of the pipeline) that bridge druggability simulations from DruGUI (left) with pharmacophore-based virtual screening (right). The names of the programs are given under each step in blue.
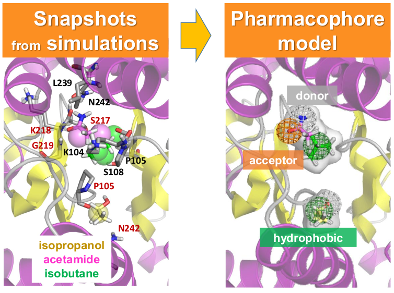
(Left) A snapshot extracted from druggability simulations for an AMPAR LBD dimer using Pharmmaker including probe poses and target conformation. Dominant binding interactions between probe and residues are shown. (Right) A pharmacophore model built based on the snapshot. One hydrogen acceptor, one donor, and two hydrophobic features were used to represent the probes.
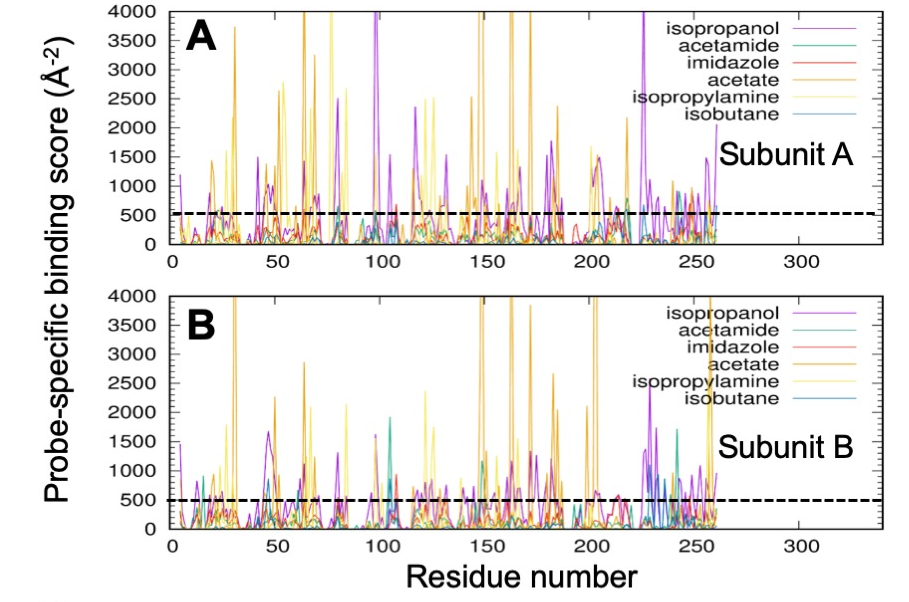
Each residue is given a binding value for each probe type, based on an inverse square distance potential. This is shown in the two graphs for the two subunits of an AMPAR LBD dimer. The dotted lines indicate a cutoff of 500, above which residues are defined as high affinity residues for a particular probe.
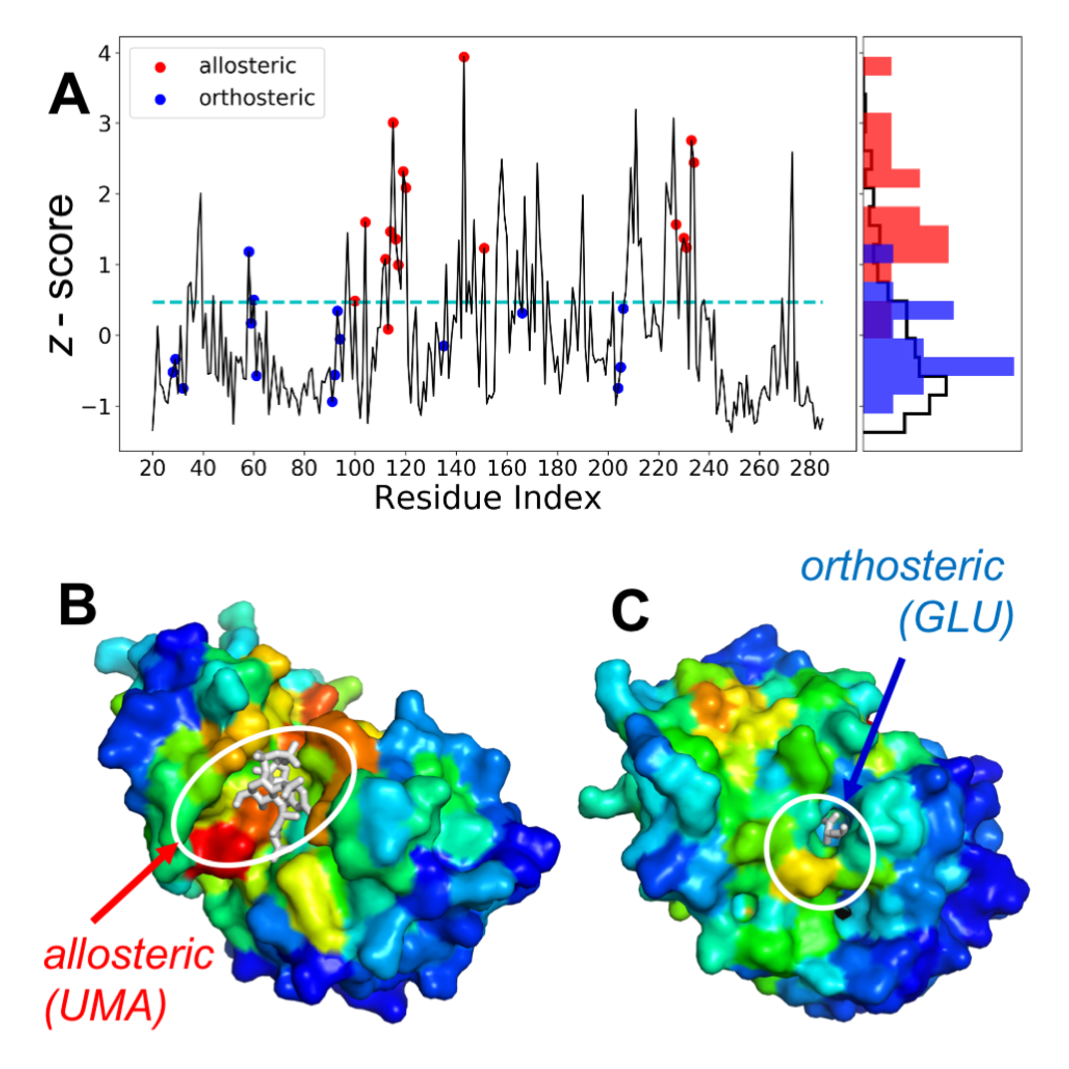
ESSA profile (A) gives a measure of the extent of frequency shift in the global modes induced by each residue. Residues (red circles) interacting with the allosteric ligand (PDB id: 2jfn) correspond to essential sites. Two differentperspectives (B-C) display color-coded by z-scores from red (highest) to blue (lowest) together with bound ligands.
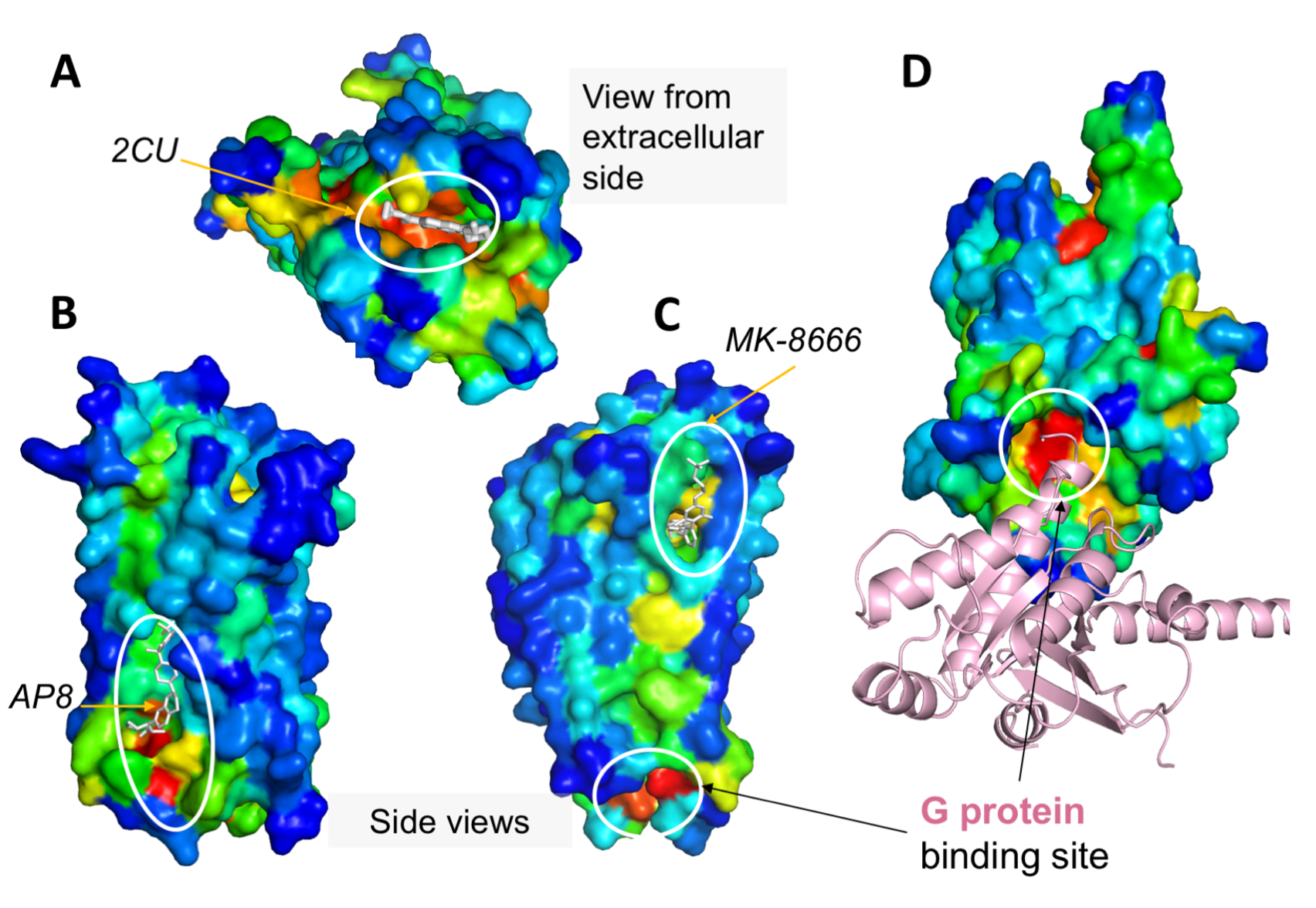
ESSA results for (A,D) muscarinic acetylcholine receptor and (B,C) free fatty acid receptor 1 GPR40. GPCRs are color-coded by the ESSA profile. Various allosteric ligand binding sites, as well as the G-protein (pink) binding site, correspond to essential or hot regions.
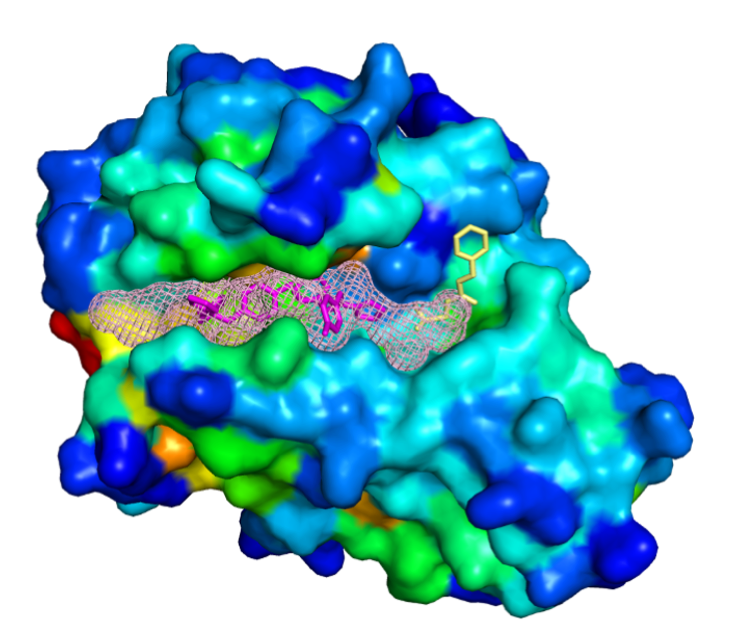
Complex structure (PDB id: 1pzo) resolved in the presence of two allosteric ligands (spatially neighboring, both shown in magenta sticks) and the orthosteric ligand (yellow sticks). The meshed surface displays the predicted allosteric pocket enclosing all ligands.
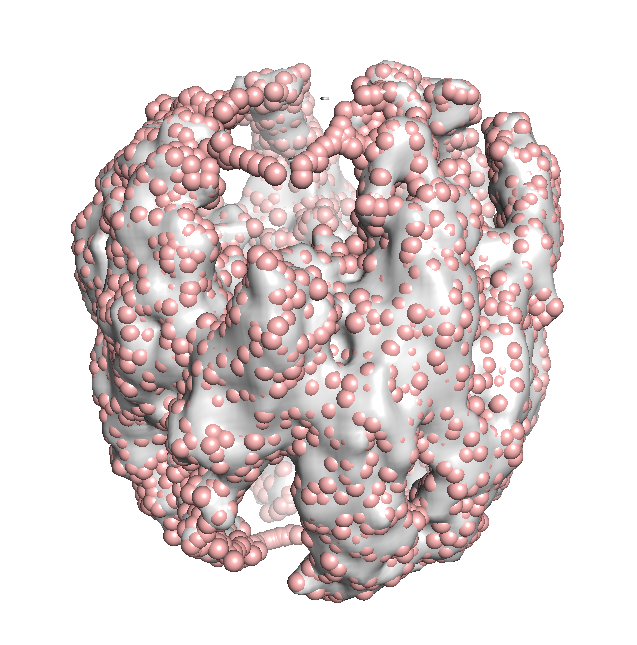
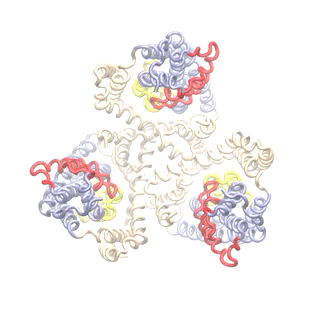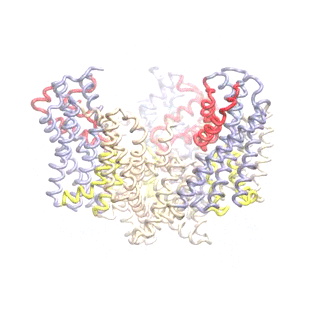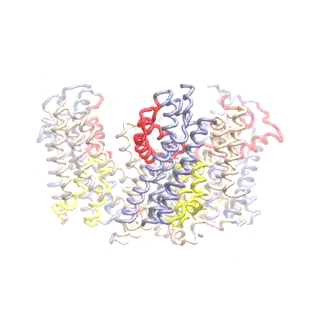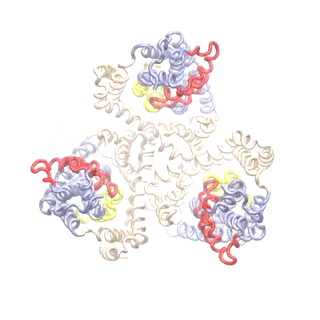
Superposition of the TRiC-AMP-PNP electron density map (EMD-1961; grey surface) and 8000 TRN nodes fitted to it (pink, spheres). This number of nodes corresponds to 1 residue/node, but higher levels of coarse-graining such as 3000 nodes works too.
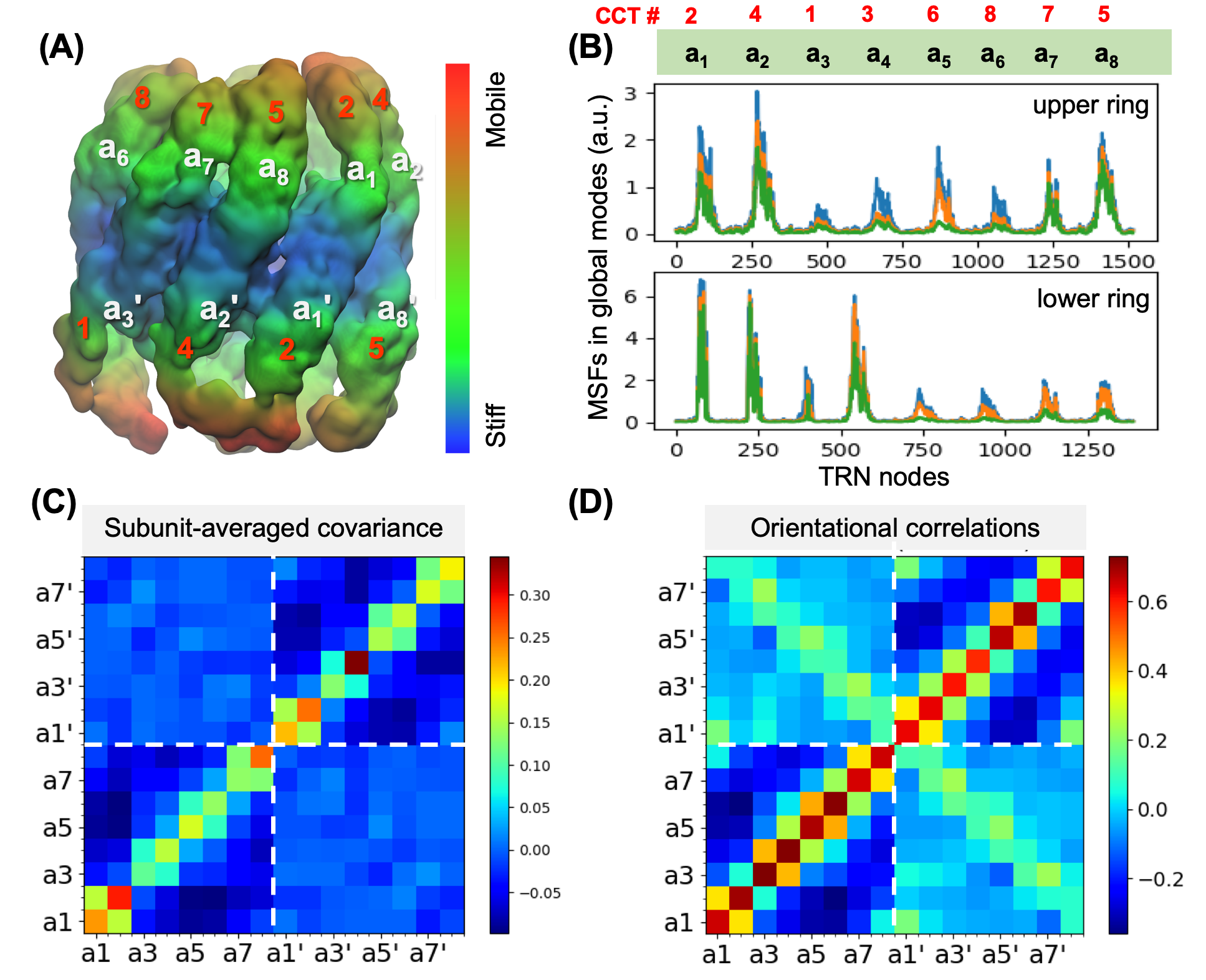
(A) Results from ANM analysis of the TRN (based on EMD-1961), displaying the architecture colour-coded by the MSFs of nodes (blue: most rigid; orange: most mobile) in the softest 20 modes. (B) MSFs of the subunits as driven by the subsets of 5 (green), 10 (orange) and 20 (blue) softest modes. (C) Covariance between the global motions of the subunits based on the softest 20 modes. (D) Orientational correlations between the global movements of the subunits.
The beads reconstructed from EMD-1961 are displaced along ANM mode 7 with an RMSD of 6 Å in both directions, revealing motions related to upper ring closure.
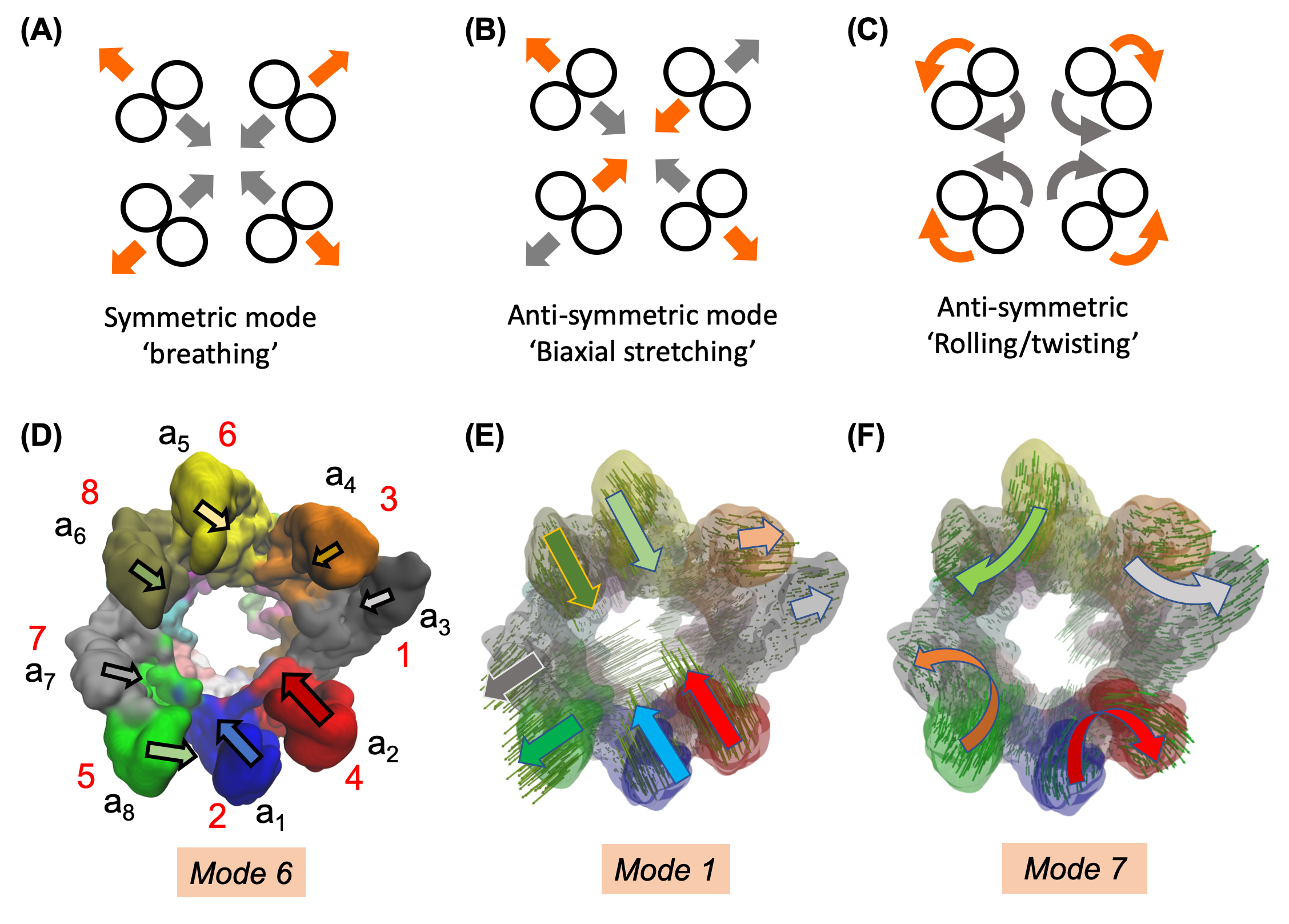
(A-C) Symmetric and anti-symmetric movements for circularly symmetric shapes. Gray and orange arrows indicate alternating motions between symmetric expansion and compression in A, stretching and contraction along orthogonal directions in B, and opposite direction rotations in C. (D-F) Modes 6, 1 and 7 of CCT/TRiC (upper ring) approximate the above motions, respectively.
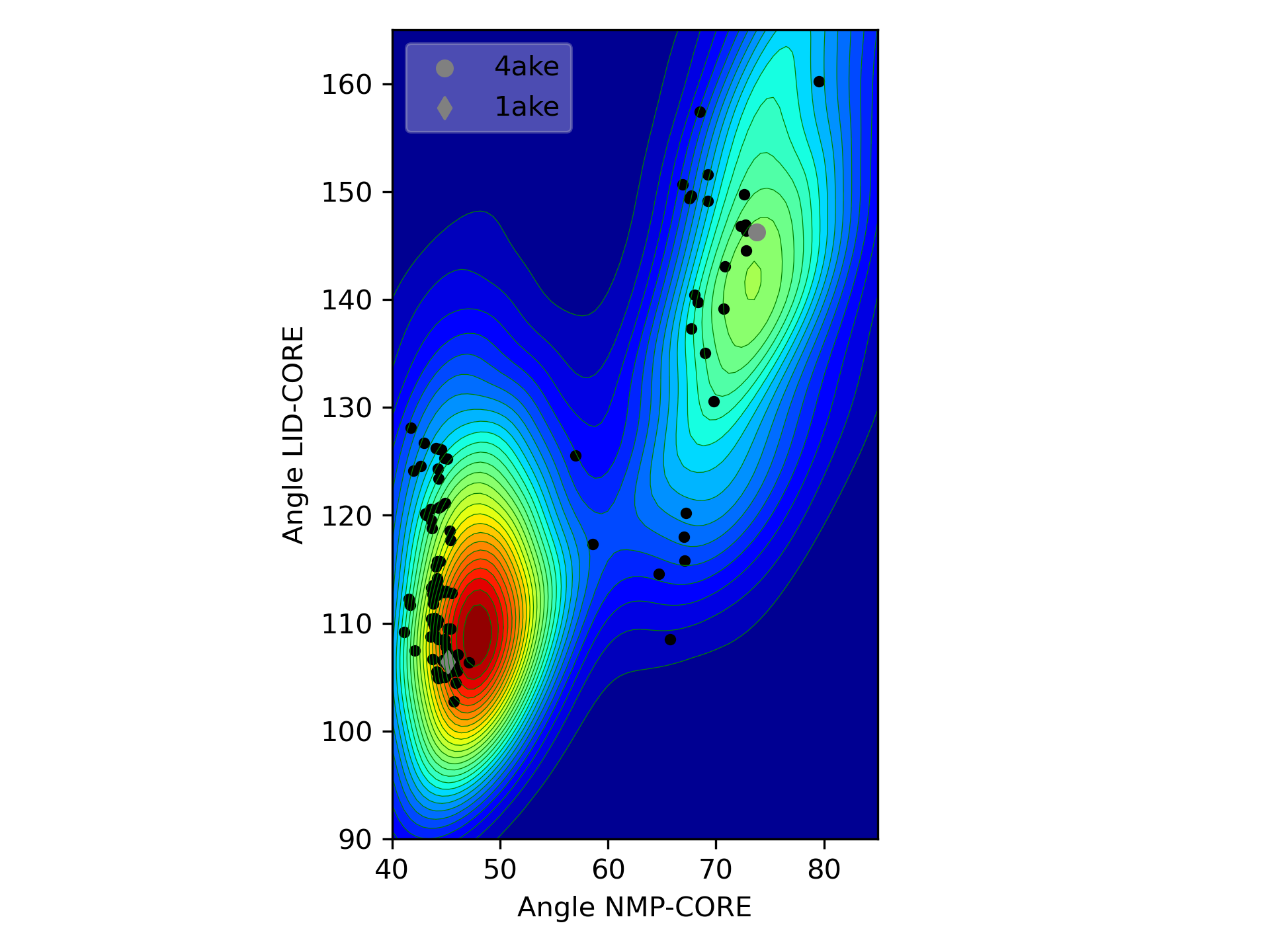
Population distribution of ClustENMD conformers shown on the angle space (LID-Core vs. NMP-Core angles) of adenylate kinase (AK), together with homologous experimental structures (black circles). Independent 5-generation runs starting from open (4ake) and closed (1ake) states of AK highlight the major minima and the populated transition states.
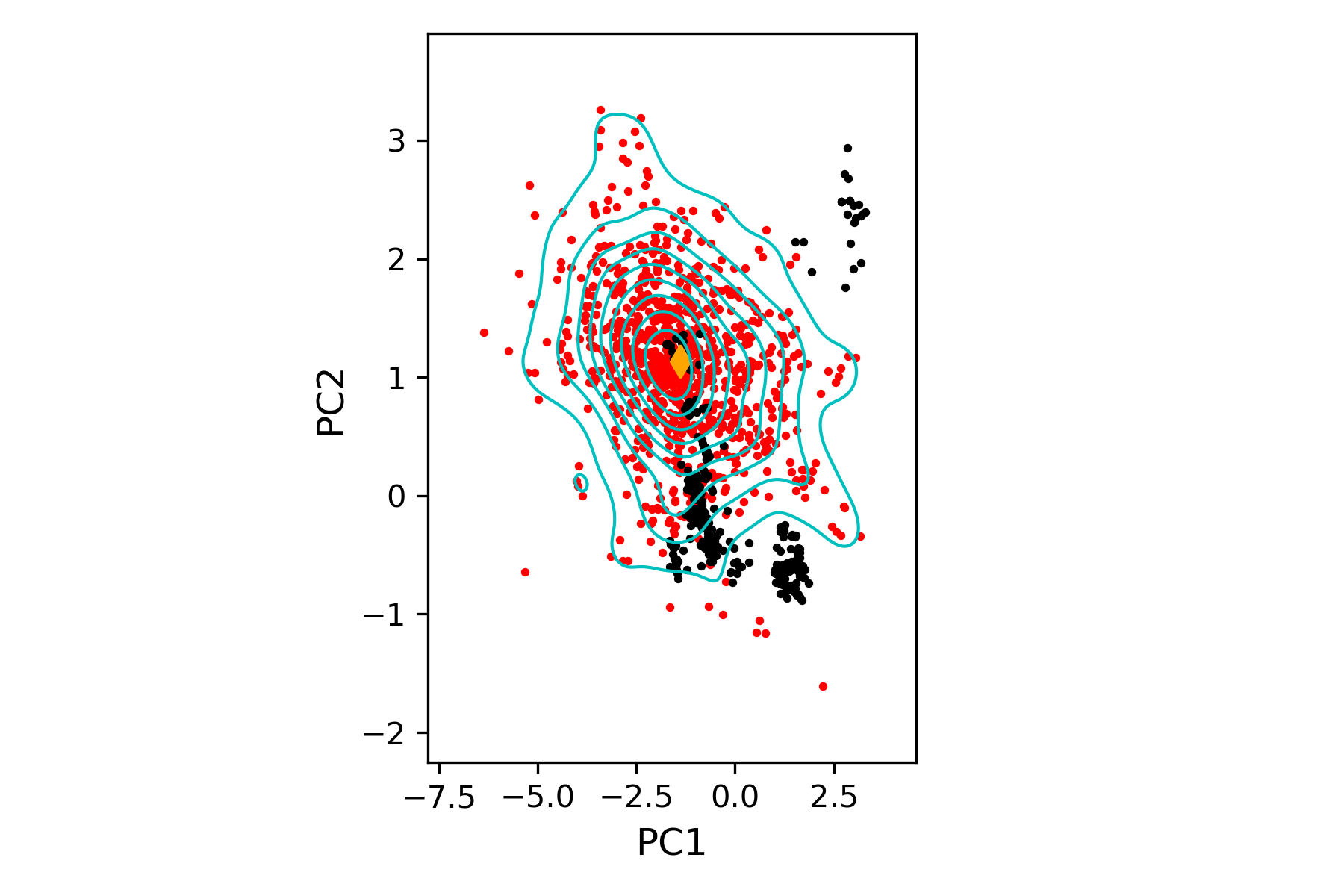
Conformational surface of HIV-1 reverse transcriptase plotted along the first two principal components (PCs) obtained from experimental structures (black circles), onto which the ClustENMD conformers (red circles, population levels in cyan) are projected.
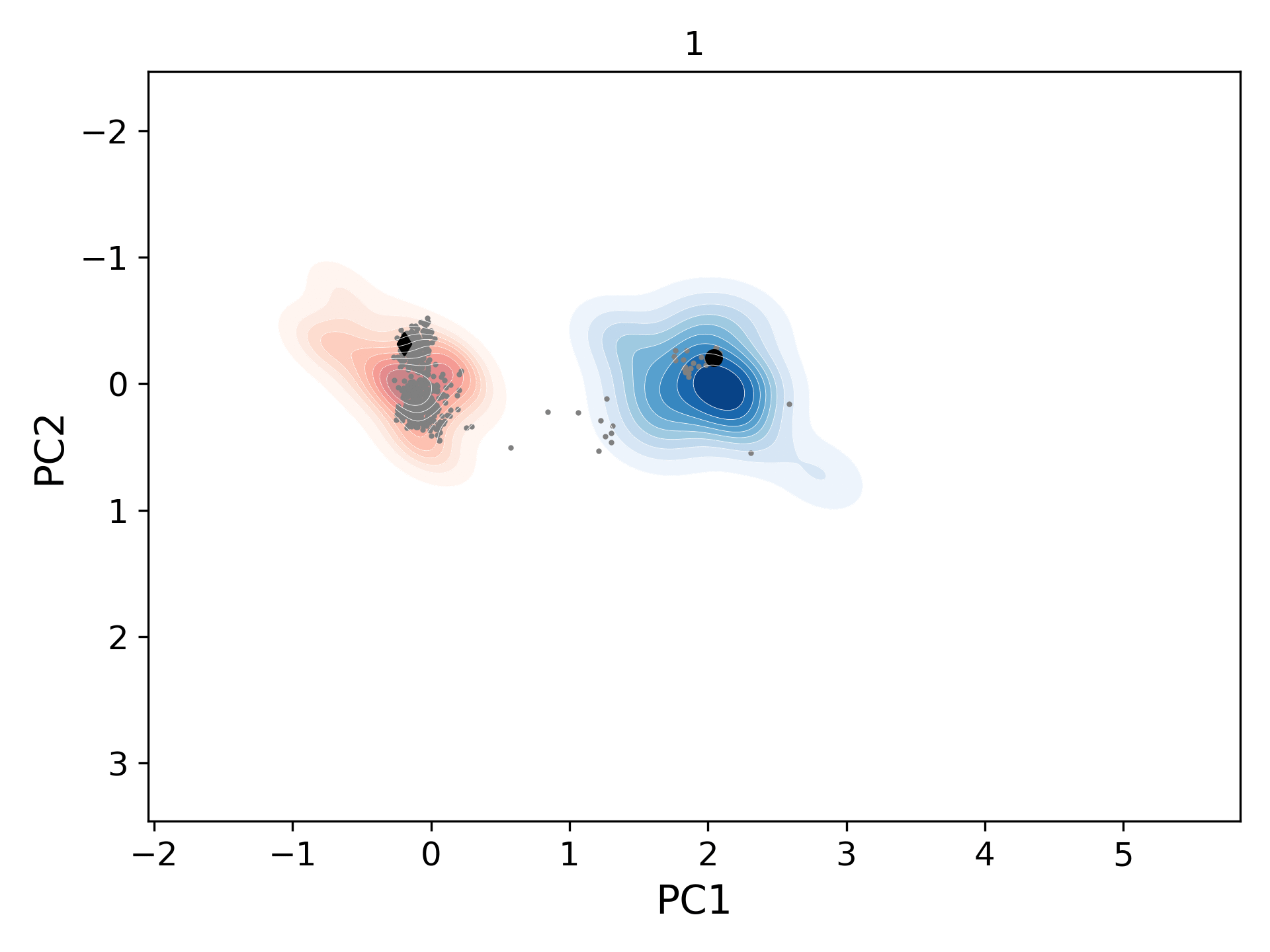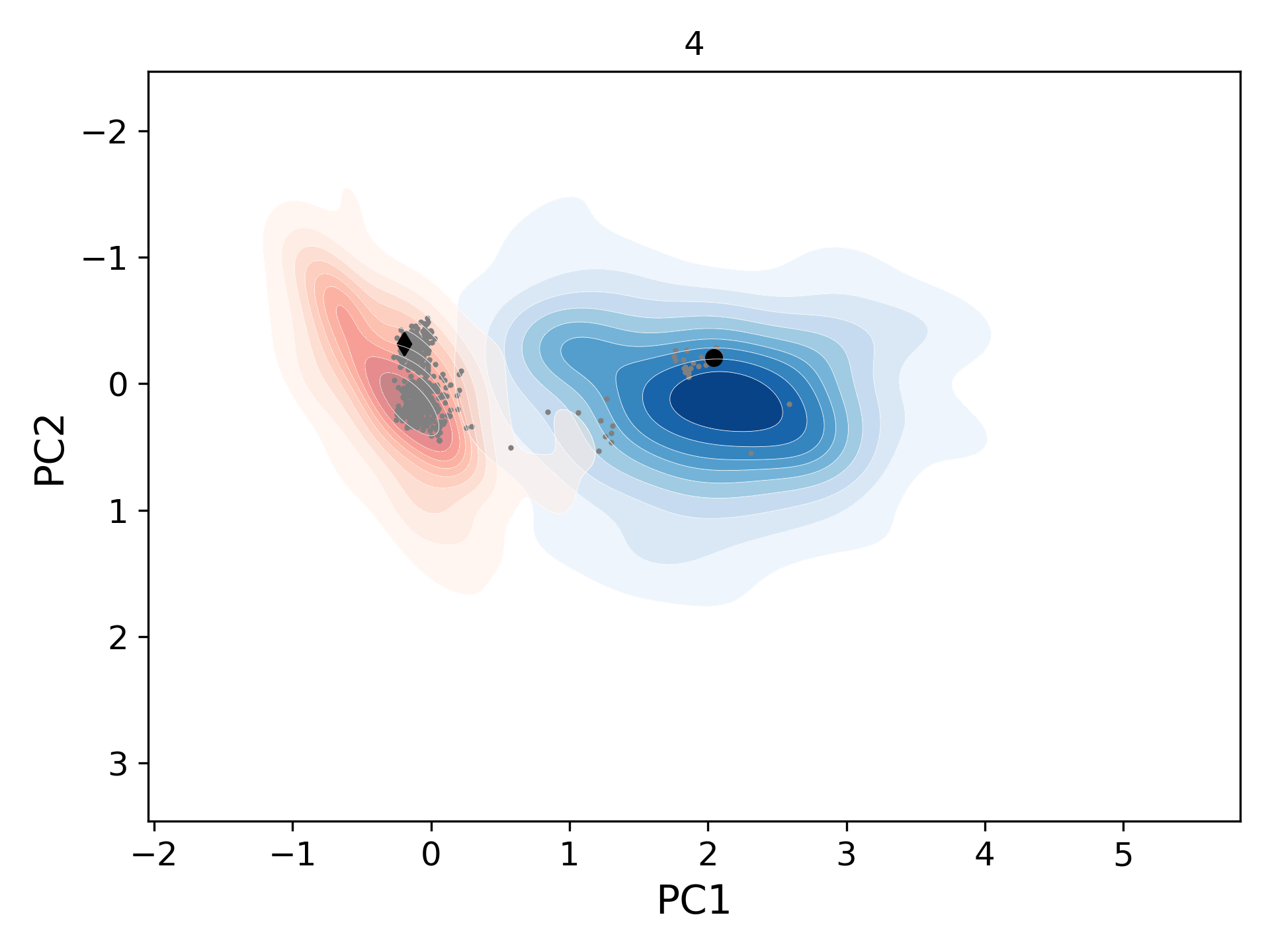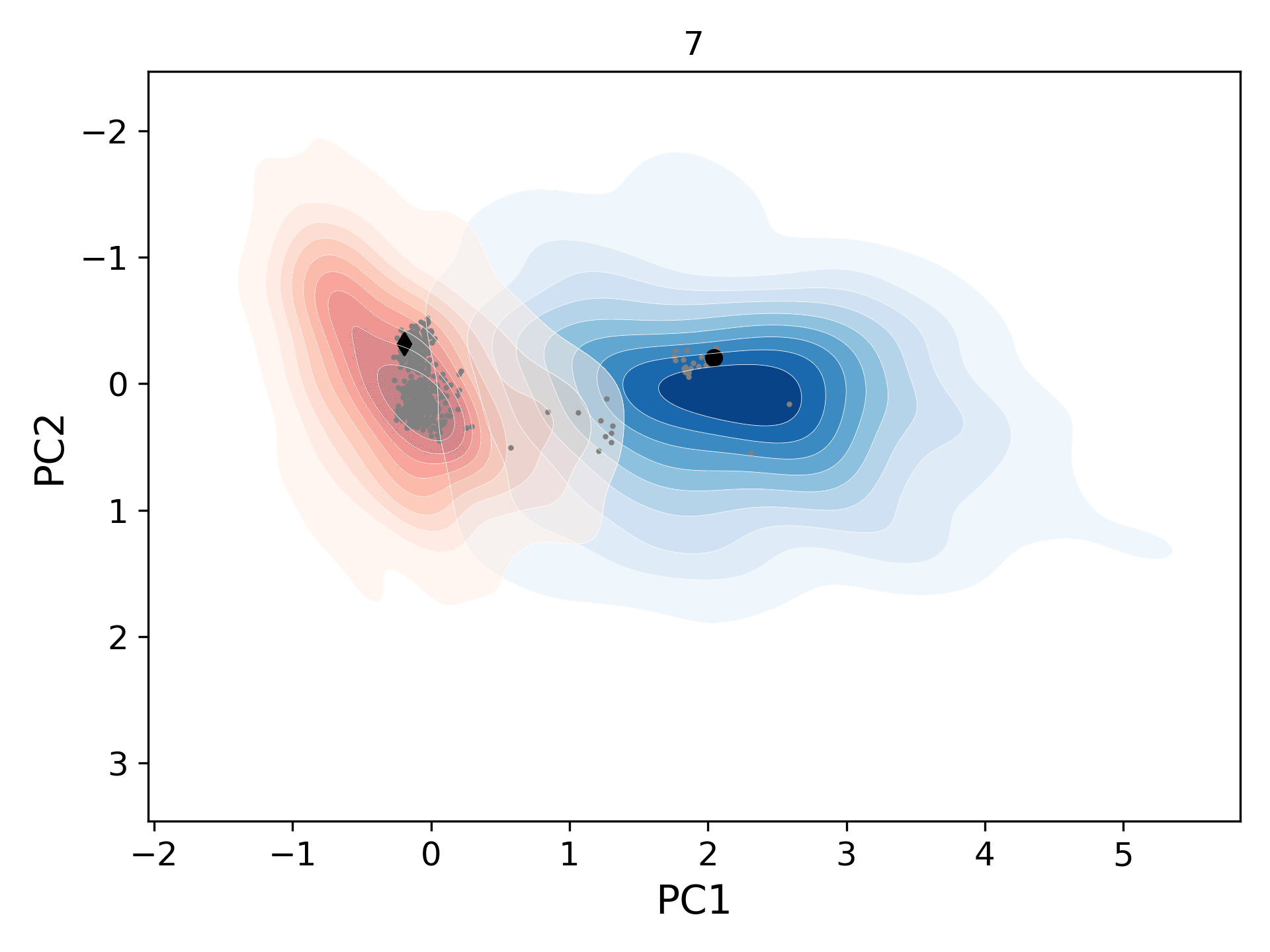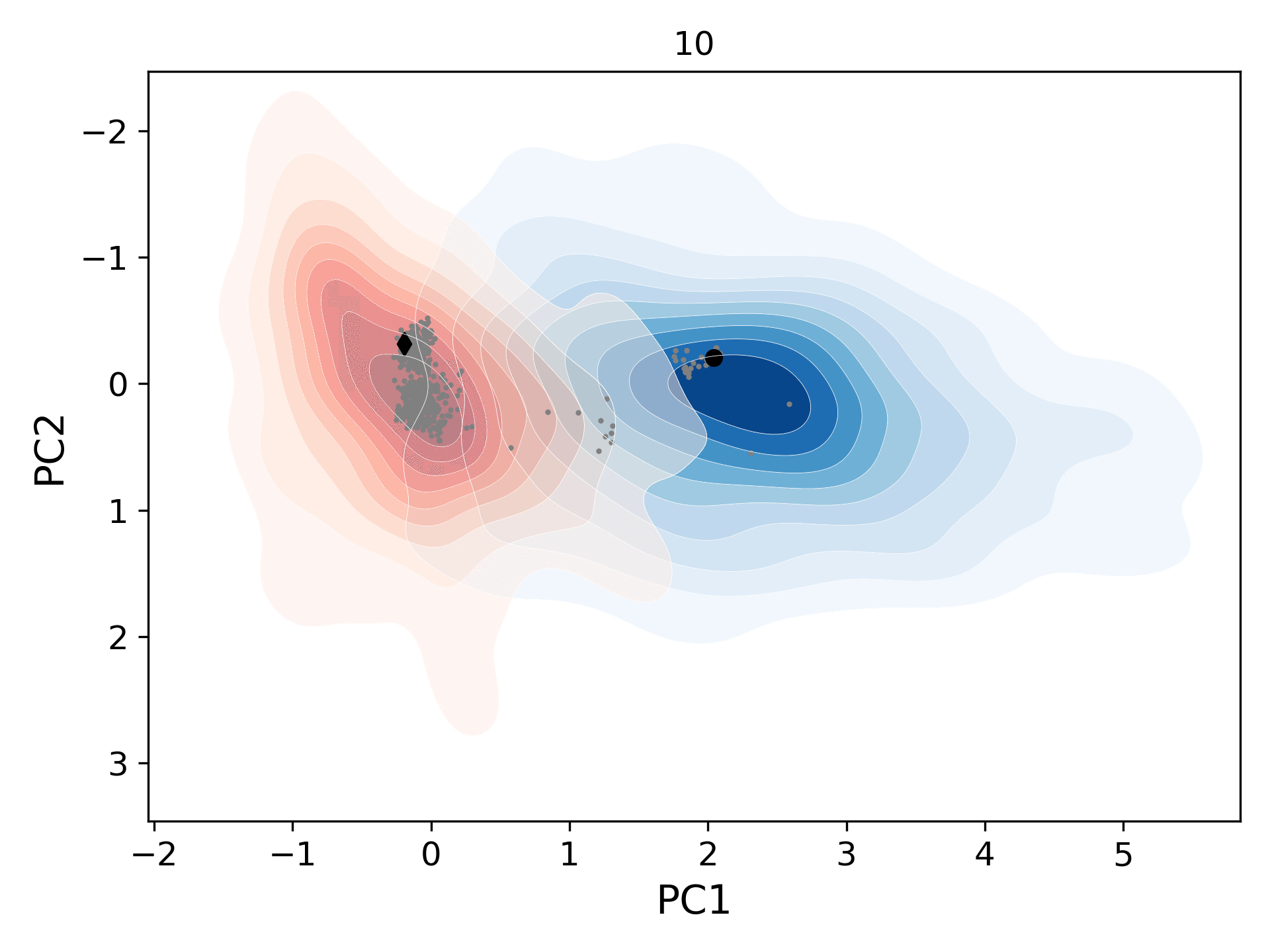
This movie shows the population distribution for successive generations (gen-1 to gen-10) of conformers sampled starting from the open (blue levels; initial structure/black circle: 1tw7) and the closed (red levels; initial structure/black diamond: 1bve) states of HIV-1 protease. As the distributions merge, they also cover the homologous experimental structures (gray circles).
CryoDy calculates the dynamics of protein complexes resolved by cryo-EM by approximating the density maps with pseudoatoms.
You can use CryoDy to
Reference
Zhang Y, Krieger J, Mikulska-Ruminska K, Kaynak B, Sorzano COS, Carazo JM, Xing J, Bahar I
State-dependent sequential allostery exhibited by chaperonin TRiC/CCT revealed by network analysis of Cryo-EM maps
2021 Prog Biophys Mol Biol 160:104-120.
ProDy is developed in Bahar Lab at the University of Pittsburgh. Click here to see a list of people who contributed to its development.
ProDy makes use of great open source software including NumPy, Pyparsing, Biopython, SciPy, and Matplotlib. Click here for details.
ProDy is open source and you can contribute to its development in many ways. See this guide for getting started.
Let us know any problems you might have by opening an issue at the tracker so that we can make ProDy better.